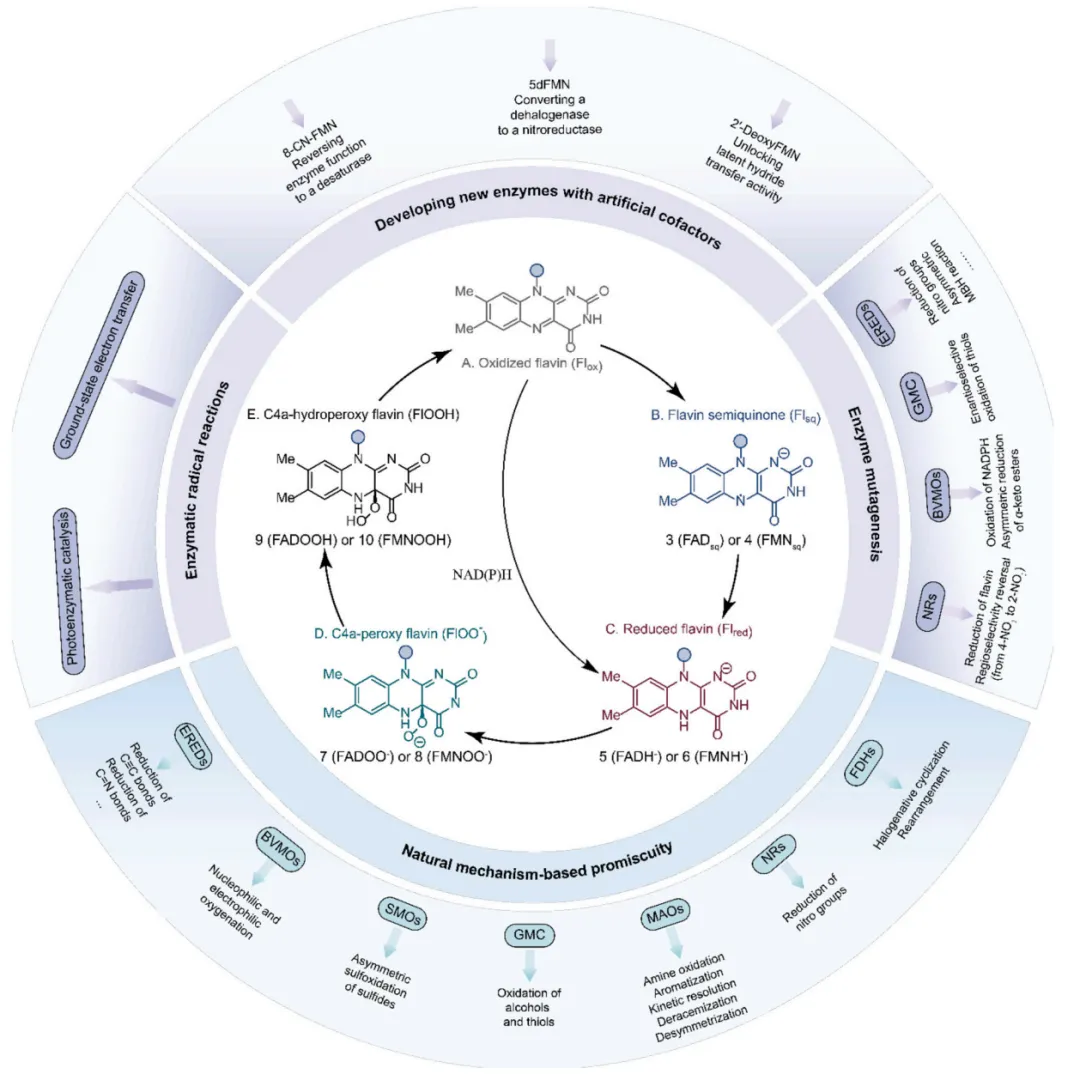
酶的催化杂泛性(Catalytic promiscuity)已成为现代生物催化领域的核心概念,它并非进化的瑕疵,而是扩展化学反应空间的巨大潜能 。在众多酶家族中,黄素依赖型酶因其辅因子——黄素单核苷酸(FMN)和黄素腺嘌呤二核苷酸(FAD)具备极其丰富的氧化还原化学性质,能够支持单双电子转移及氧气活化,从而在催化杂泛性方面占据独特地位 。近期的一项全面综述将黄素依赖型酶的反应性系统地划分为“基于自然机制的杂泛性”与“区别于自然机制的杂泛性”两大类,其底层的机制图谱清晰地展示了不同酶家族如何通过调节黄素状态来实现反应路径的多样性分支(图1) 。
图1. 黄素依赖型酶反应多样性的机制基础
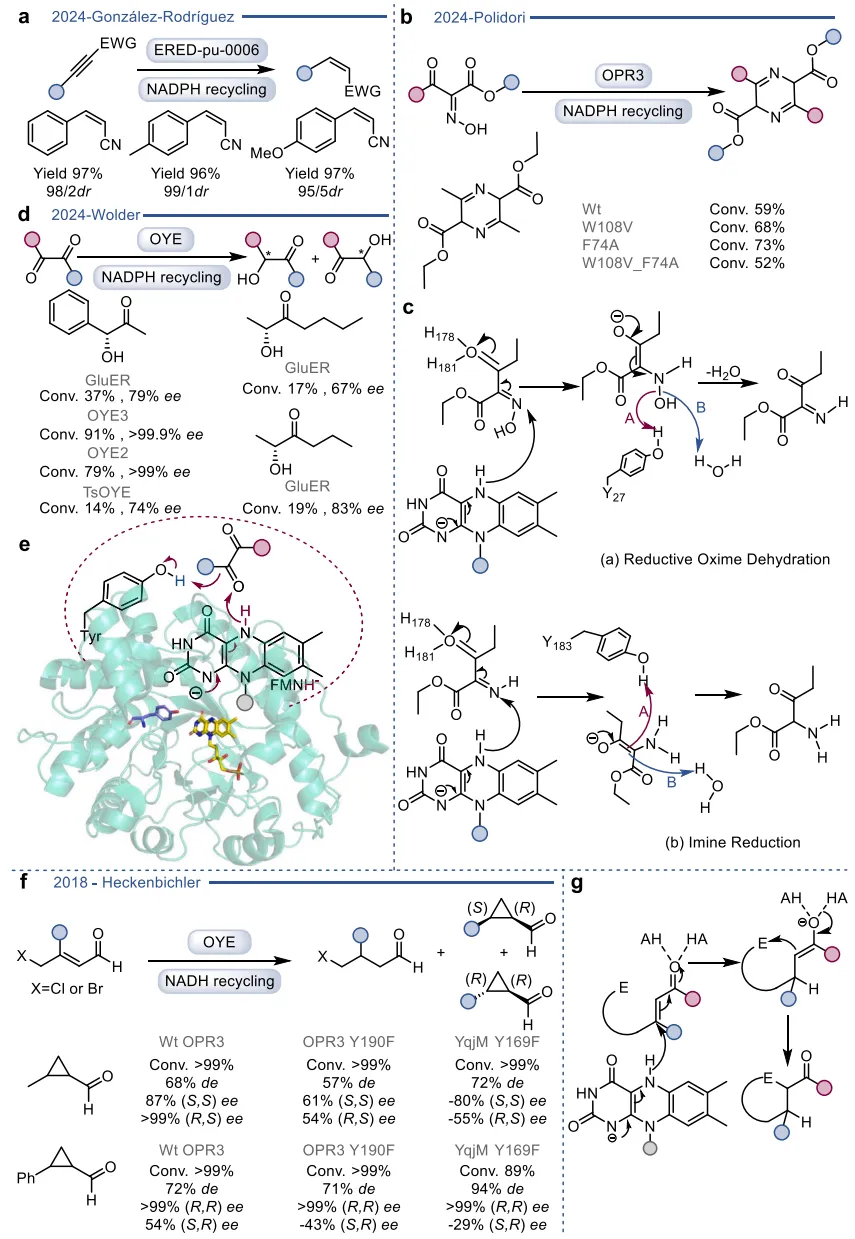
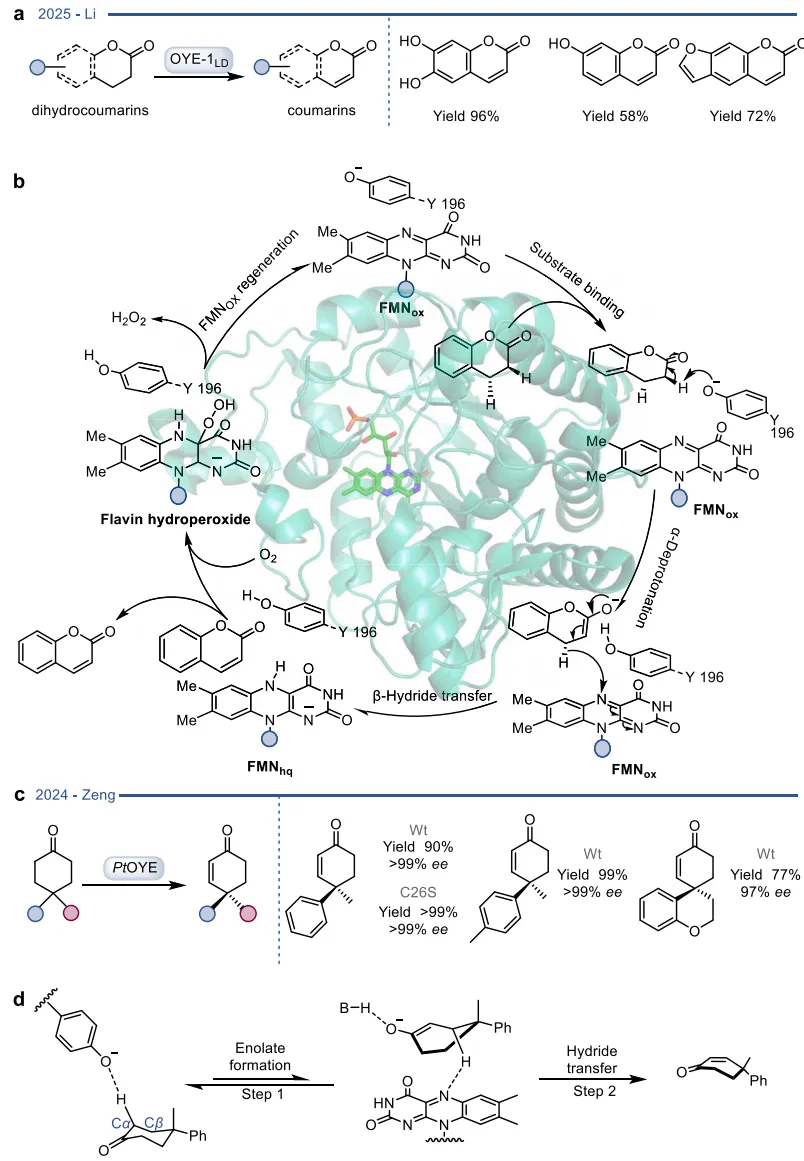
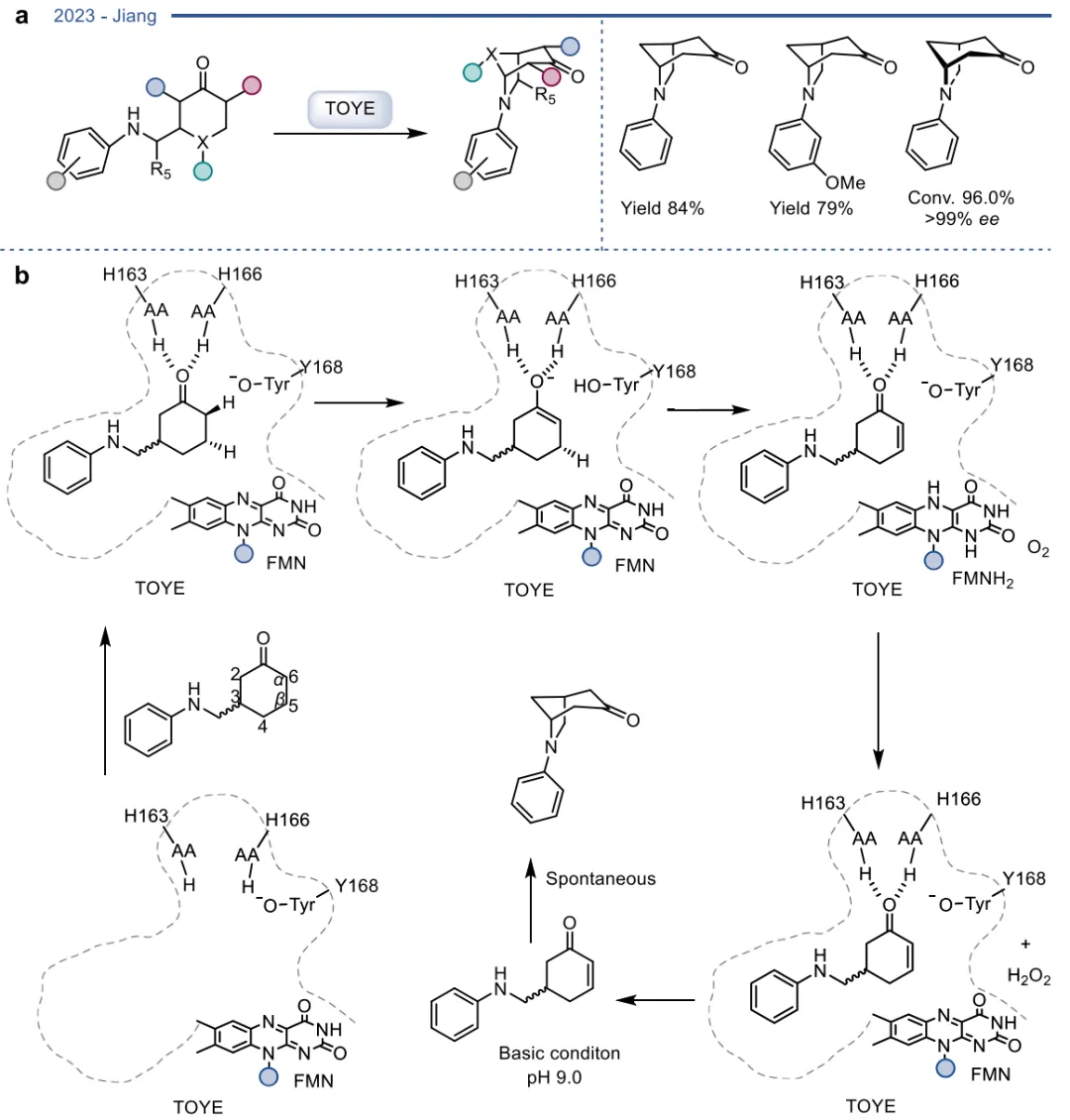
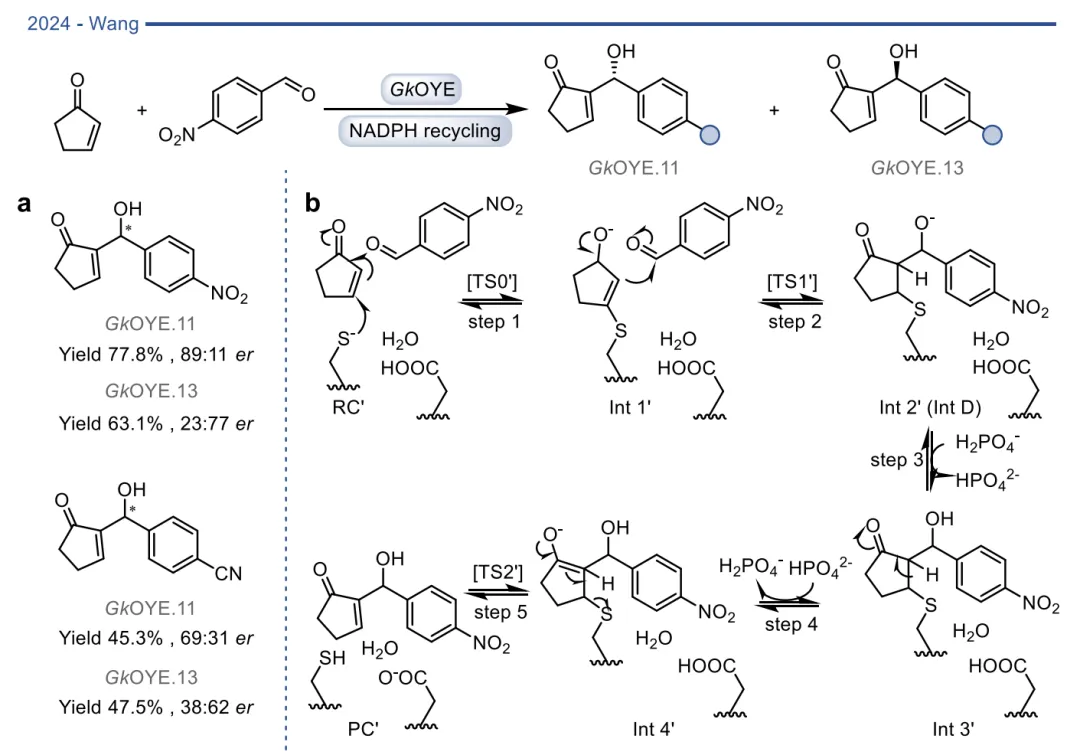
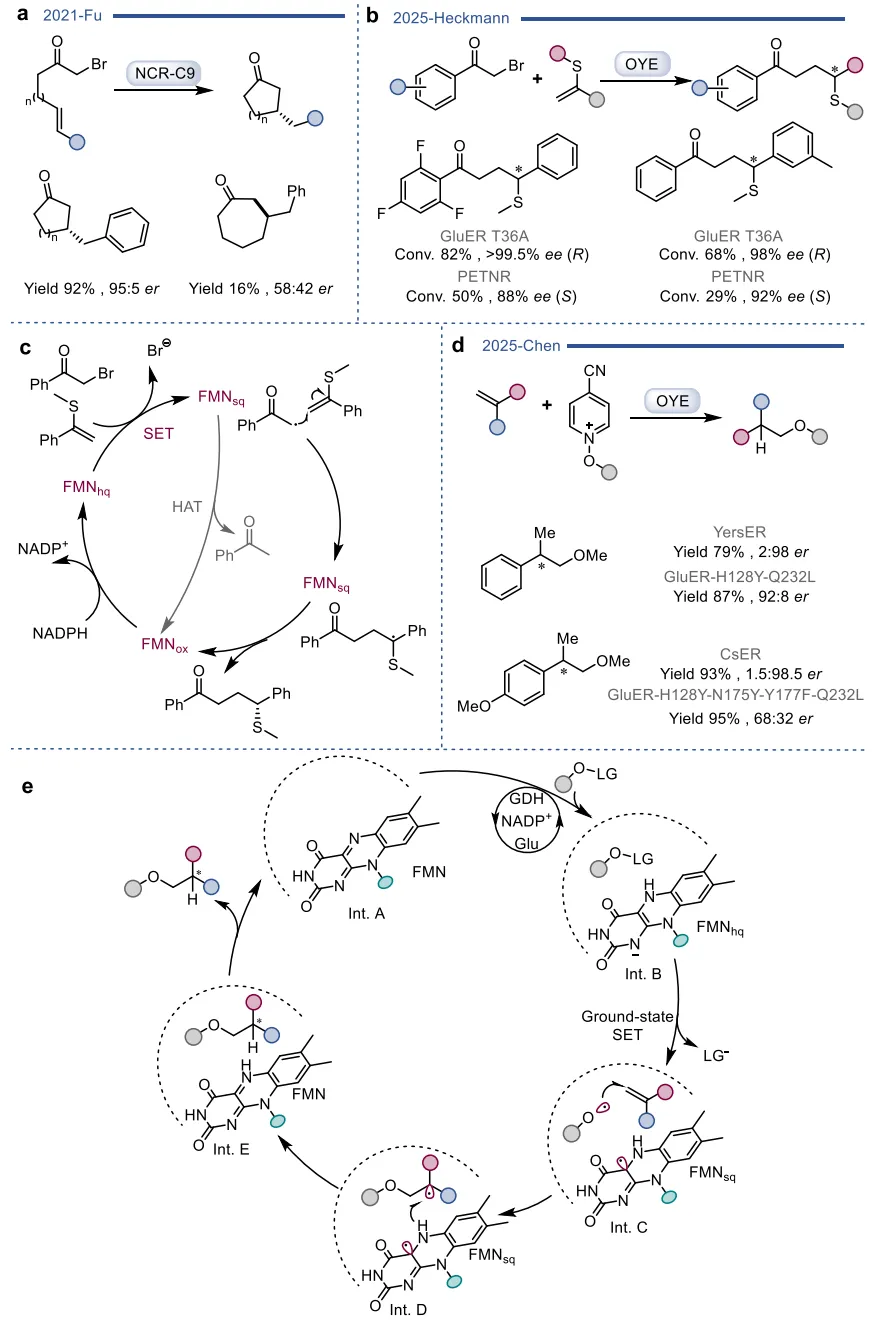
在基于自然机制的杂泛性范畴内,酶在保留其固有催化框架的前提下,被重新定向以处理新底物或催化非天然反应 。烯还原酶(EREDs,特别是老黄酶OYEs)是这一策略的典型代表。其天然机制是通过“乒乓”机制利用NAD(P)H提供的氢负离子立体选择性地还原活化烯烃 。在此基础上,这类酶的还原范围被大幅拓展,可用于不对称还原碳碳叁键、碳氮双键(如将肟还原为胺)以及碳氧双键,甚至能够利用还原性环化实现碳碳键的偶联(图3)。更令人瞩目的是,在缺乏NAD(P)H辅因子时,这类还原酶能够发生功能“反转”,利用氧气作为末端氧化剂,高效催化脱氢去饱和氧化(图4)、脱氢环化(图5)以及脱氢胺化与脱氢芳构化(图6) 。此外,特定的OYEs甚至能在不依赖黄素氧化还原化学的情况下,通过酸碱催化介导碳碳双键的异构化(图7) 。
图3. 基于自然机制实现OYE催化反应多样性的实例
图4. OYE催化的氧化去饱和反应实例
图5. TOYE介导的脱氢环化反应
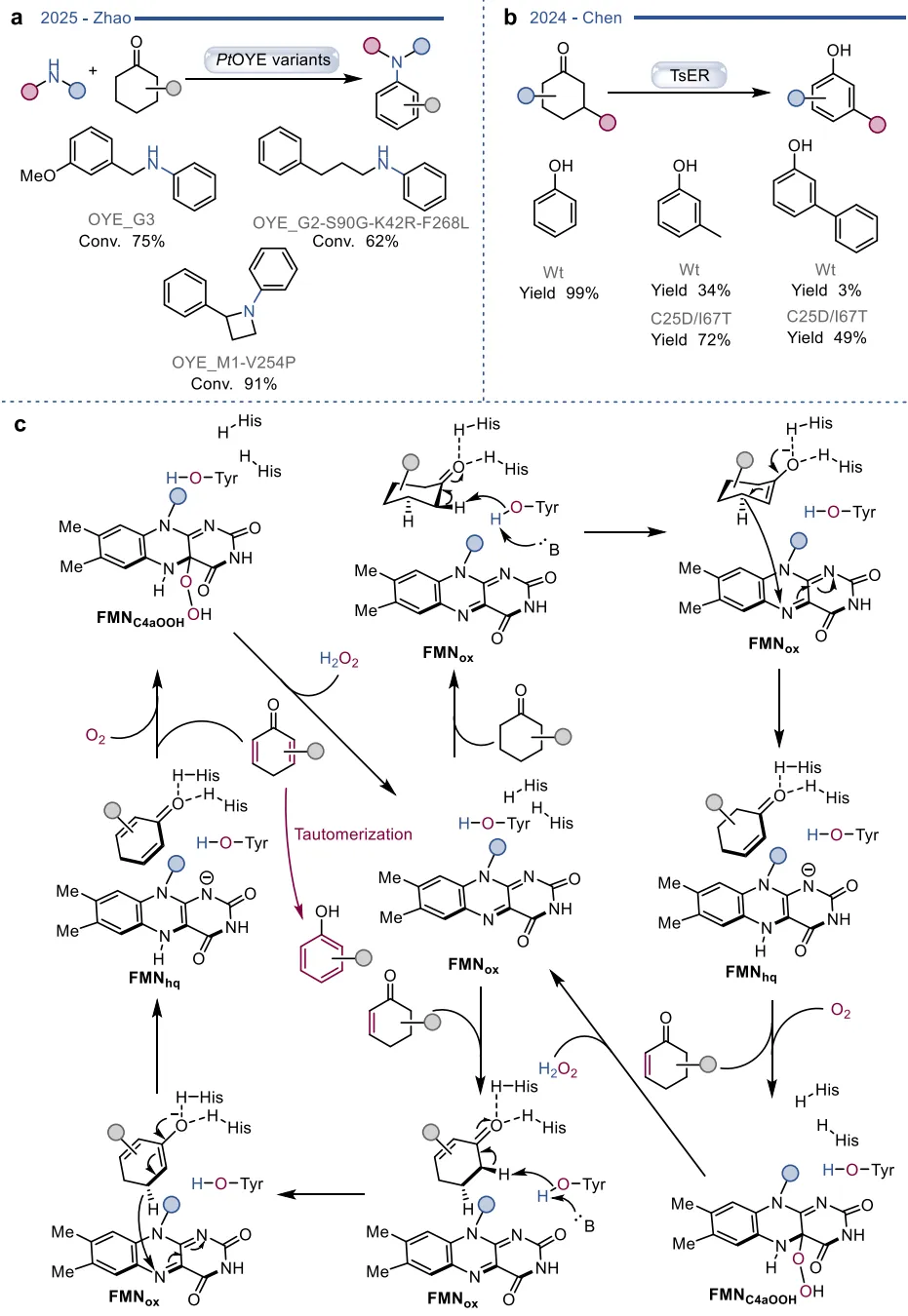
图6. PtOYE催化的脱氢胺化反应与TsER介导的脱氢芳构化反应
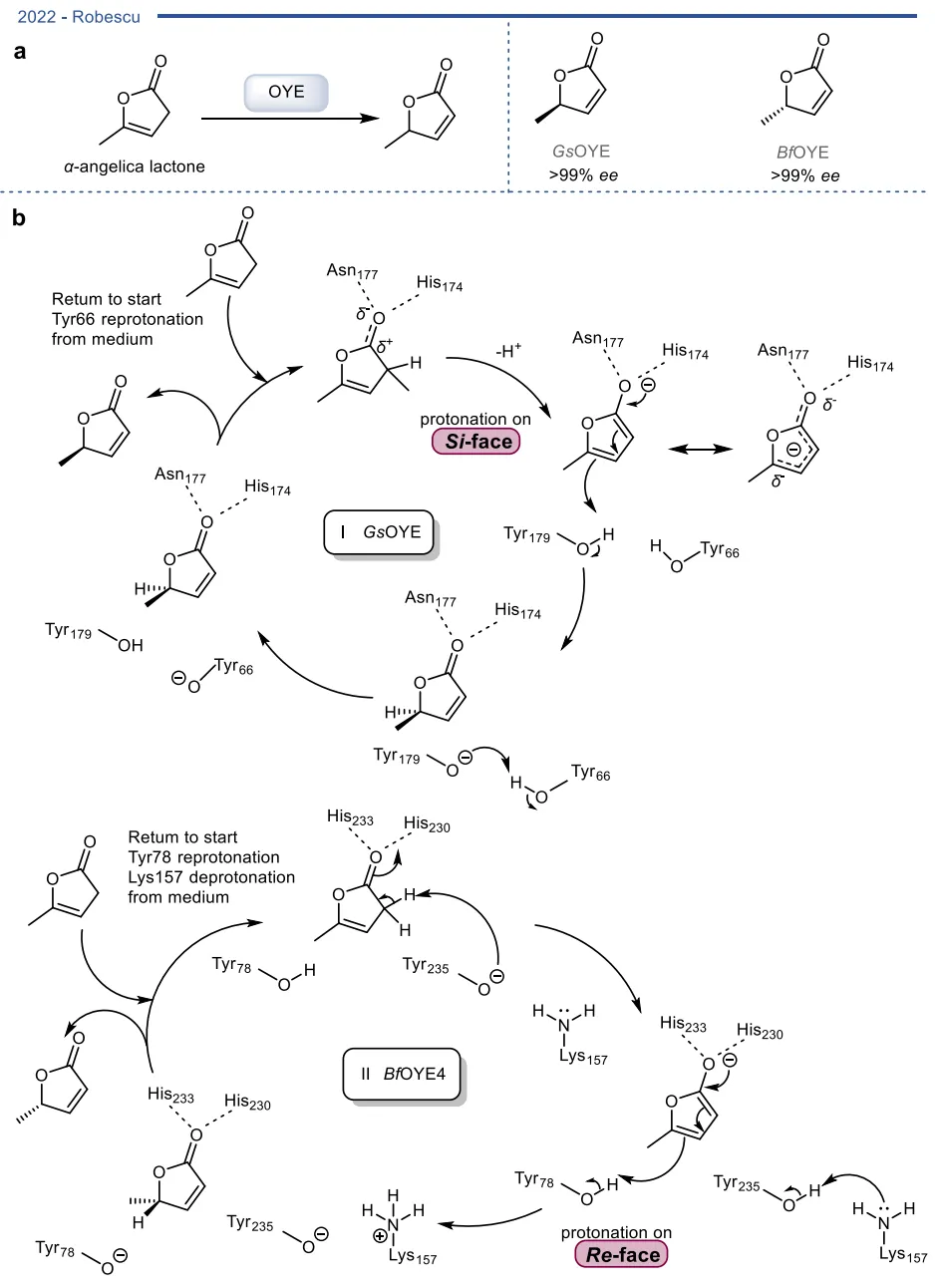
图7. 两种不同OYE介导的碳碳双键对映互补异构化反应
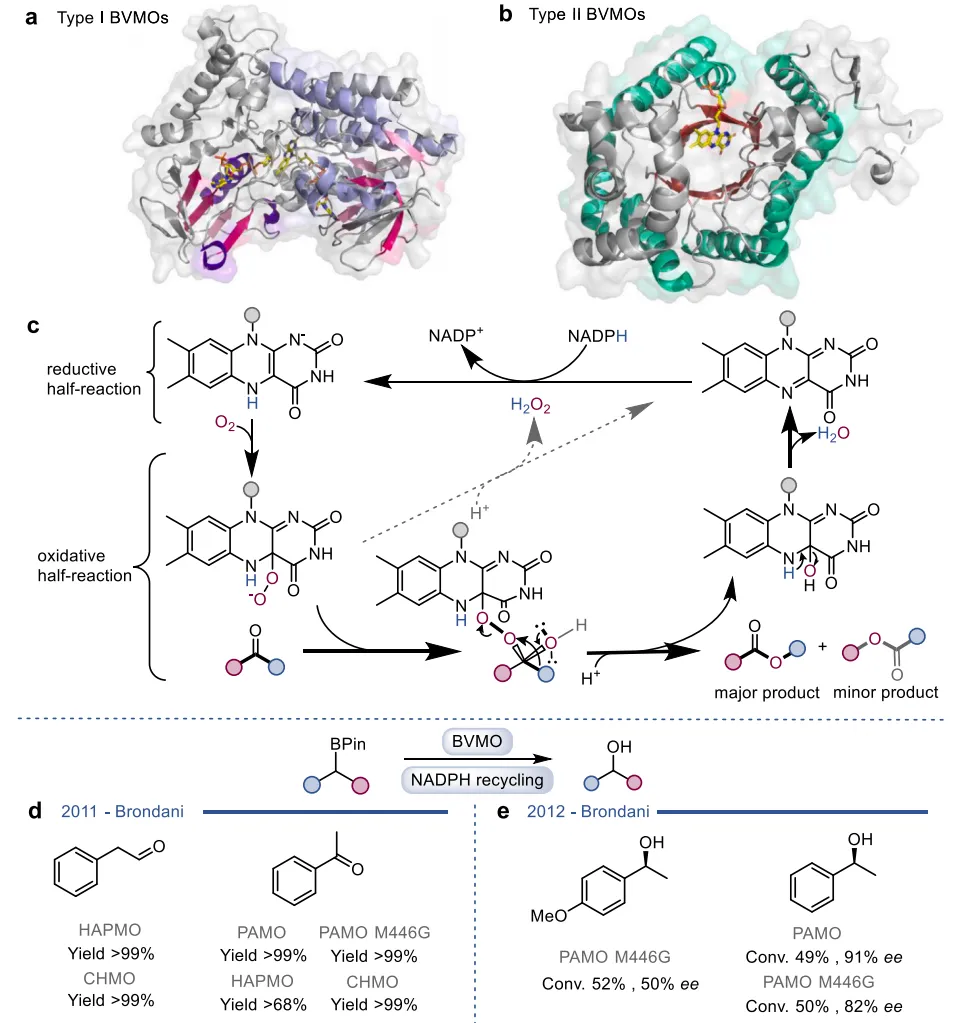
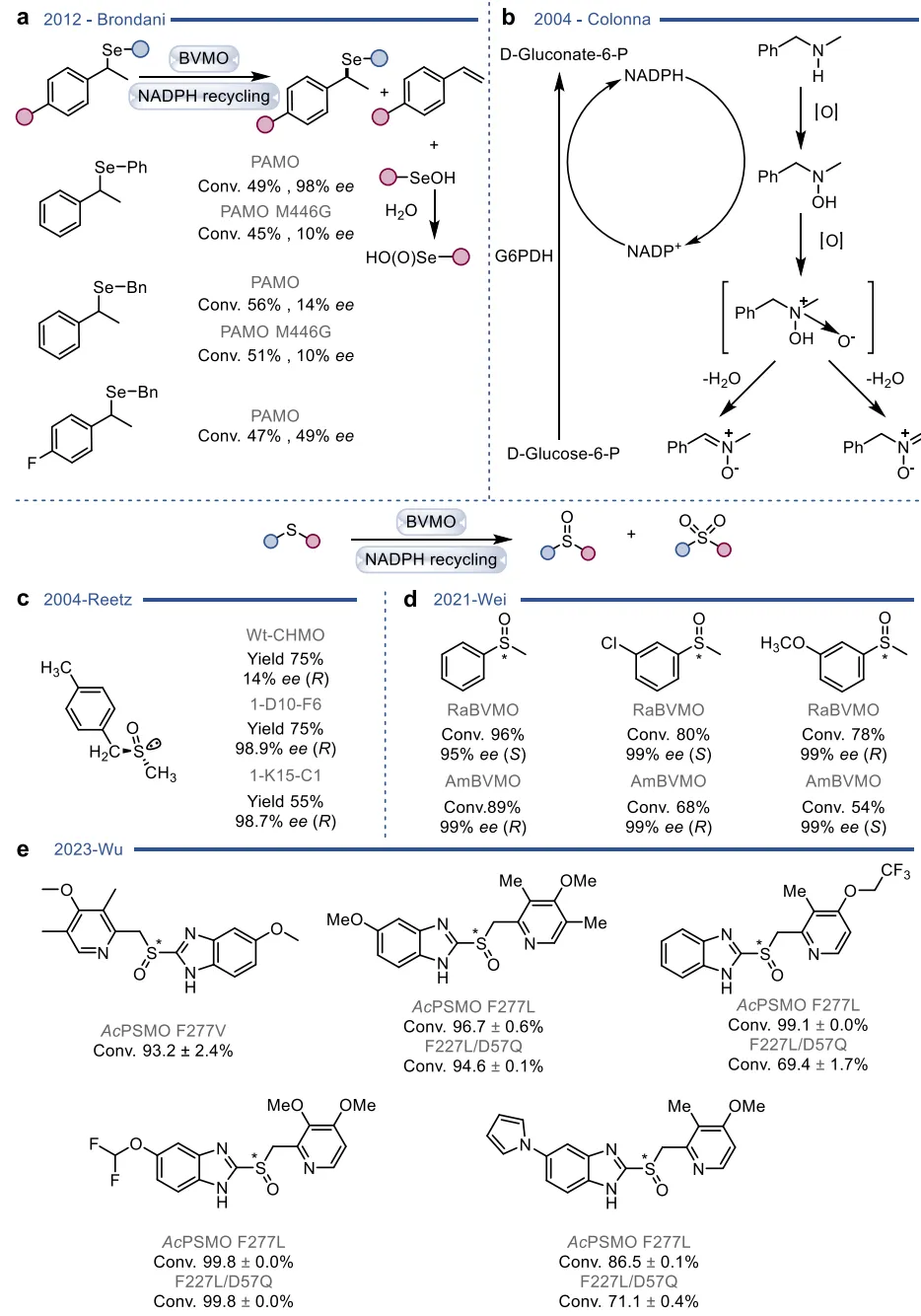
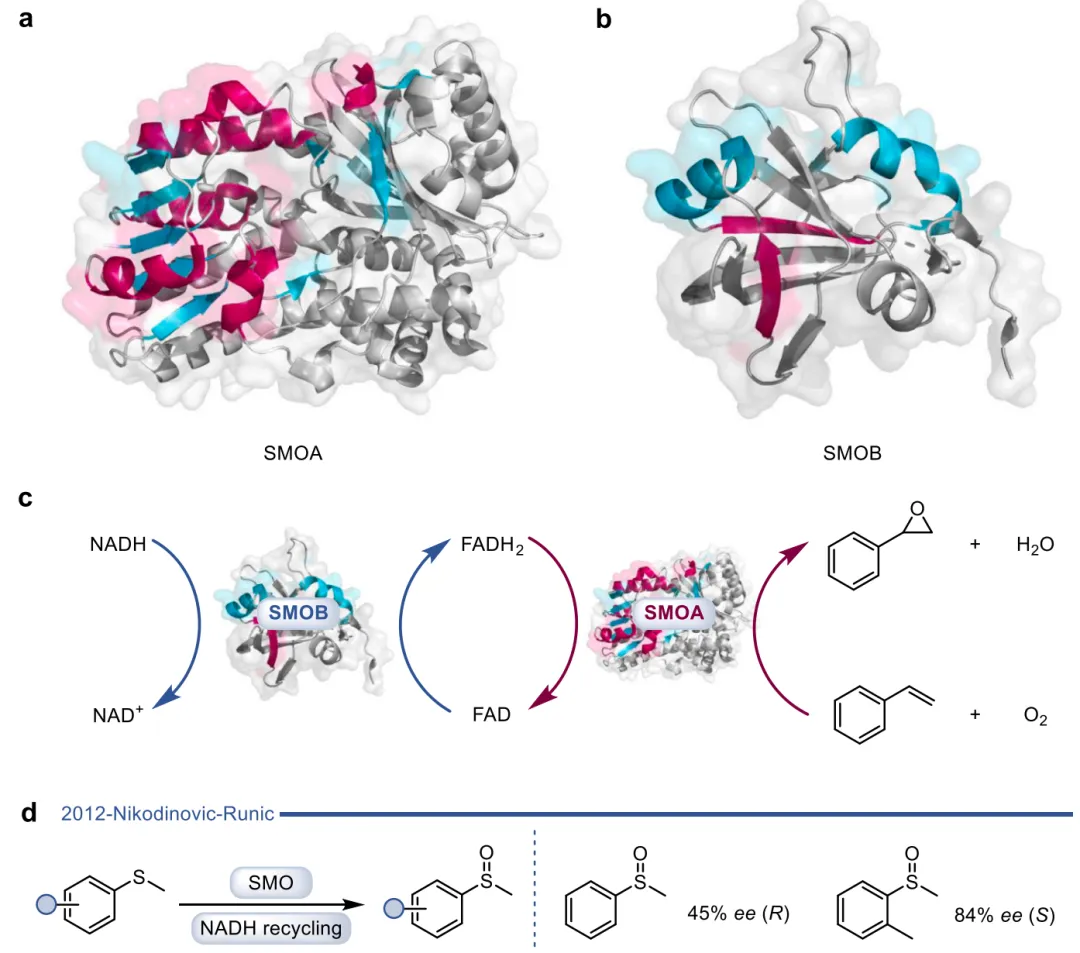
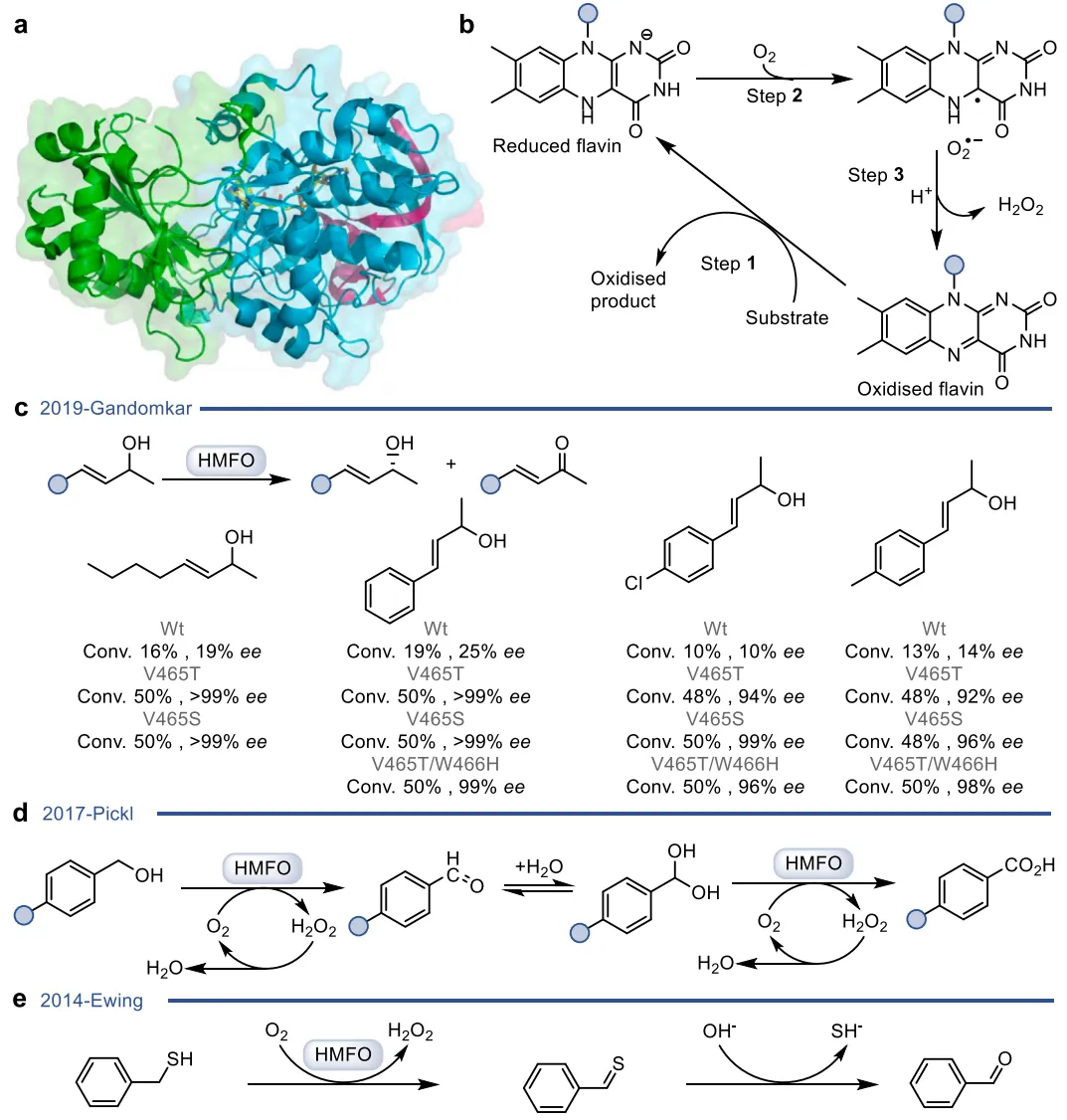
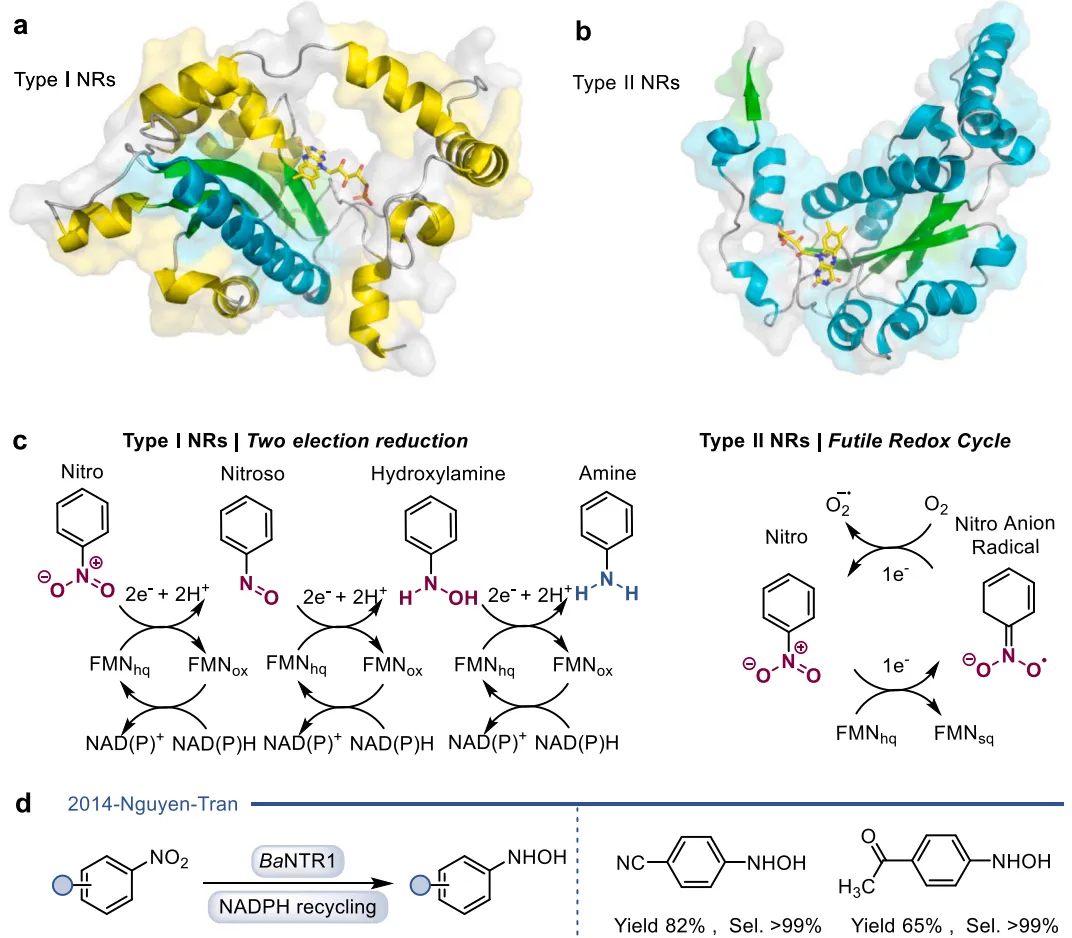
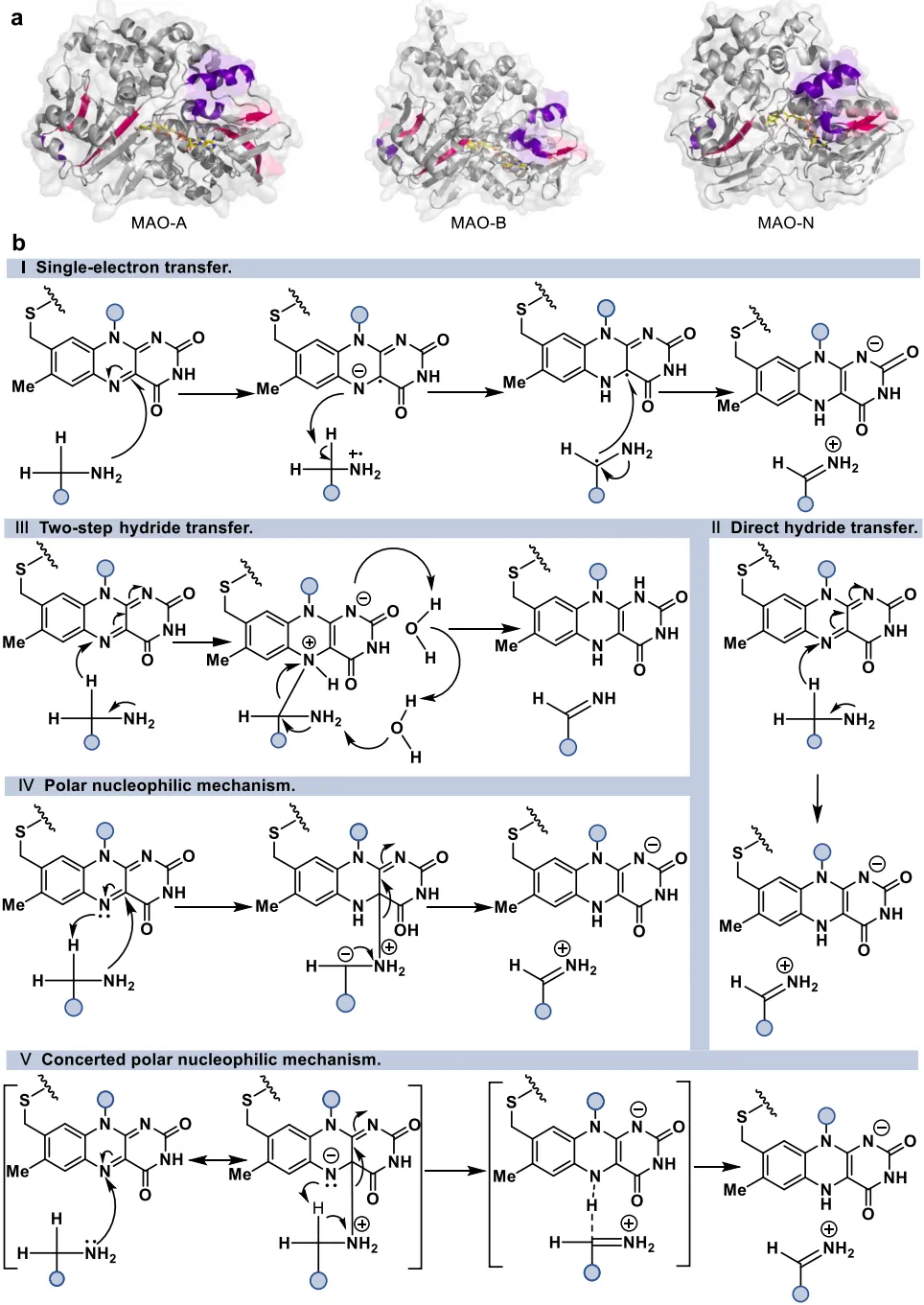
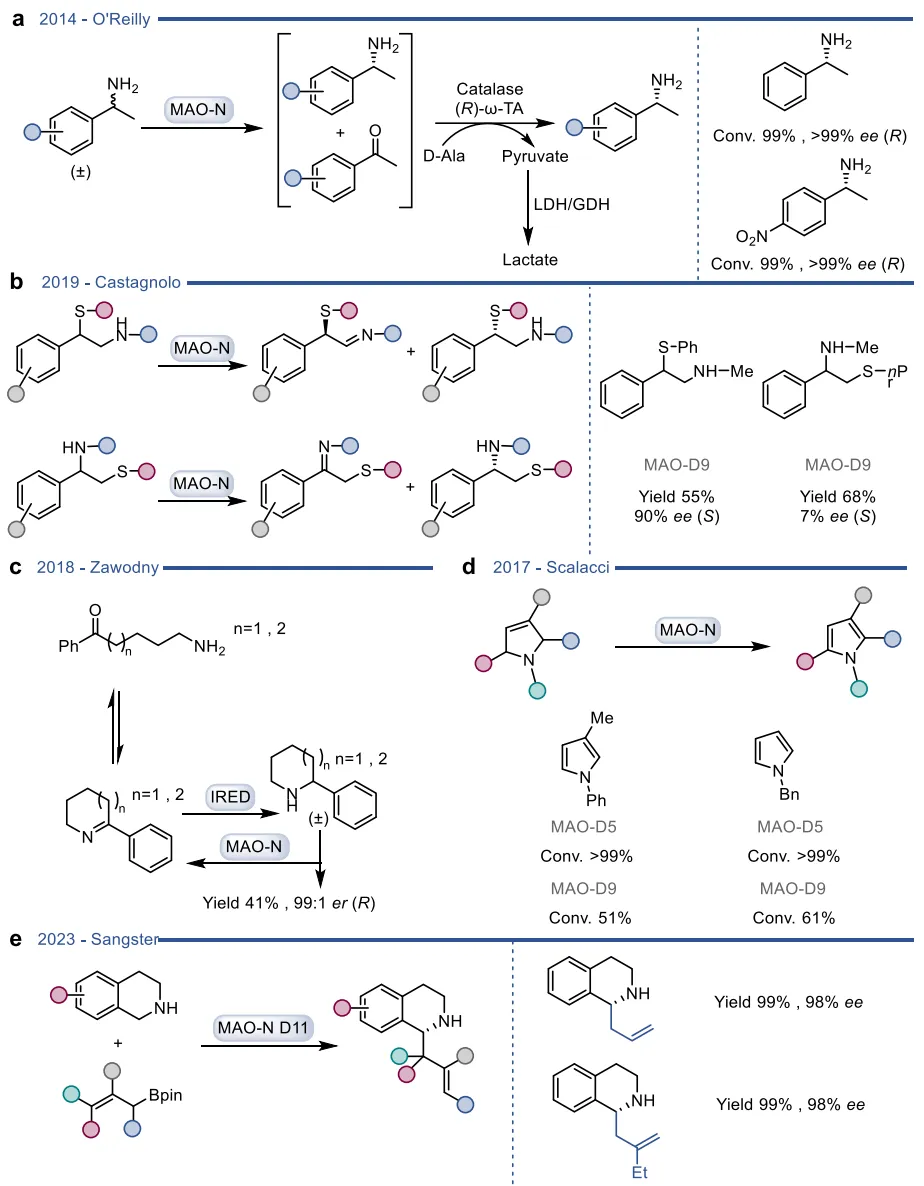
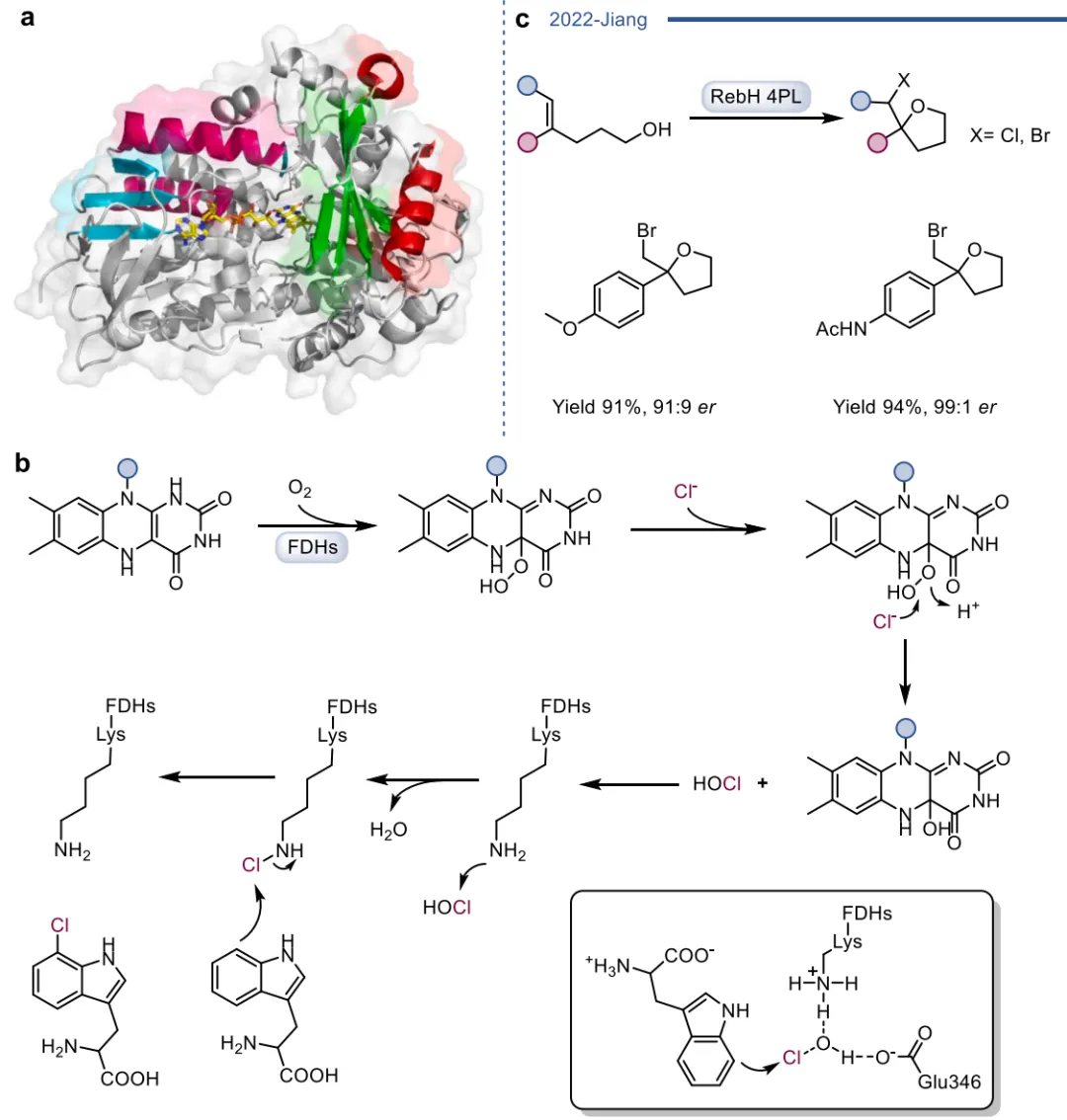
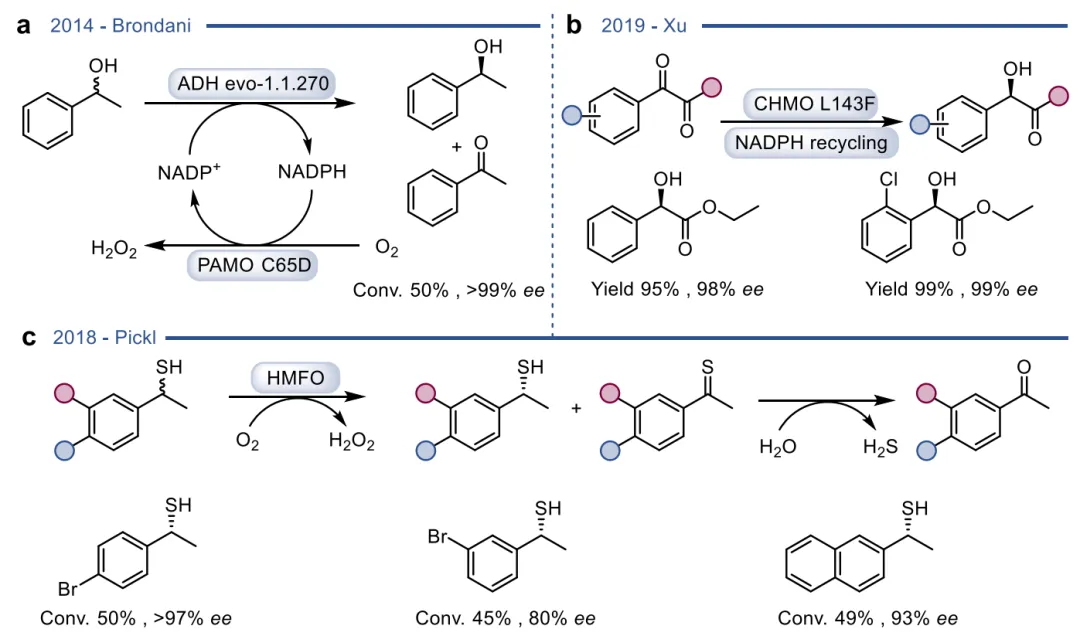
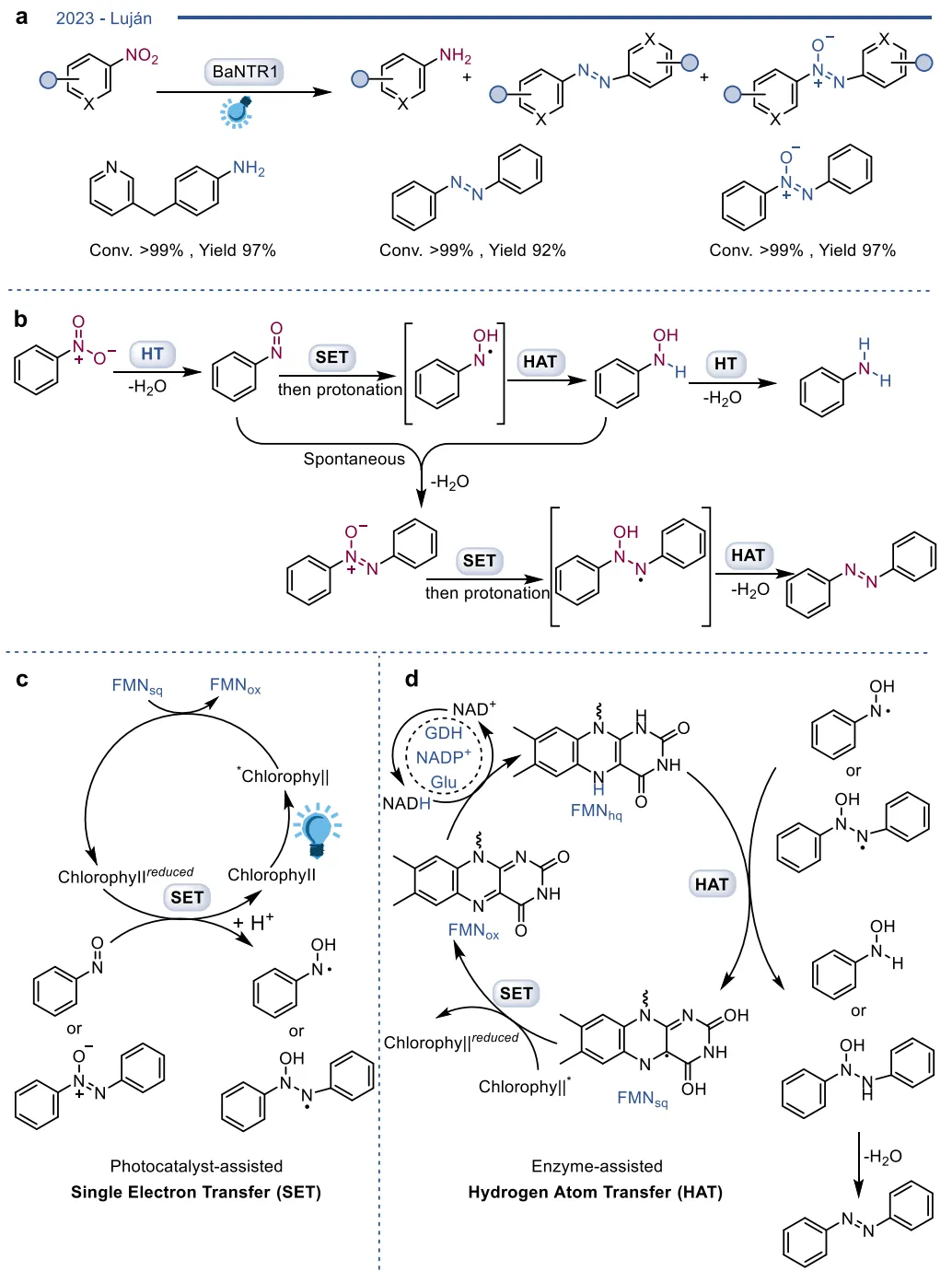
黄素蛋白单加氧酶(FPMOs)同样展示了机制的横向延伸。经典的拜耳-维立格单加氧酶(BVMOs)利用过氧黄素中间体催化底物的亲核加成,利用这一固有能力,BVMOs可进一步实现非天然的碳硼键氧化(图8),以及针对硒、氮、硫等杂原子的极高对映选择性亲电氧化(图9) 。苯乙烯单加氧酶(SMOs)也利用类似的黄素氧活化机制实现了芳基硫醚的不对称硫氧化(图10) 。在其他酶家族中,GMC型氧化酶(如HMFO)的底物谱被拓宽至大位阻醇类及硫醇的氧化(图11) ;硝基还原酶(NRs)通过精确的电子转移控制,实现了向N-芳基羟胺的高选择性分步还原(图12) ;单胺氧化酶(MAOs)的复杂氧化机制(图13)被成功应用于消旋胺的动力学拆分、去对称化及氧化芳构化(图14) ;黄素依赖型卤化酶(FDHs)则通过亲电卤化机制催化了不对称的卤内酯化与复杂的碳碳键重排(图15) 。
图8. I型和II型拜耳-维立格单加氧酶(BVMOs)的晶体结构与经典氧化机制,及其向碳硼键氧化的催化拓展
图9. BVMO催化碳硒键、碳氮键氧化
图10. 苯乙烯单加氧酶(SMOs)双组分的代表性晶体结构、经典环氧化催化机制,以及其向碳硫键氧化的催化延伸
图11. HMFO的晶体结构与天然反应机制,及其催化仲烯丙醇、苄醇和硫醇氧化的拓展应用
图12. I型和II型硝基还原酶(NRs)的晶体结构、经典还原反应机制,以及BaNTR1高选择性还原含吸电子基团的硝基芳烃生成N-芳基羟胺的过程
图13. 单胺氧化酶(MAO-A、MAO-B、MAO-N)的晶体结构及其五种可能存在的催化机制模型
图14. MAO-N催化胺类官能团化的应用实例
图15. 黄素依赖型卤化酶(FDH)的晶体结构、卤化机制以及烯烃的卤代醚化反应
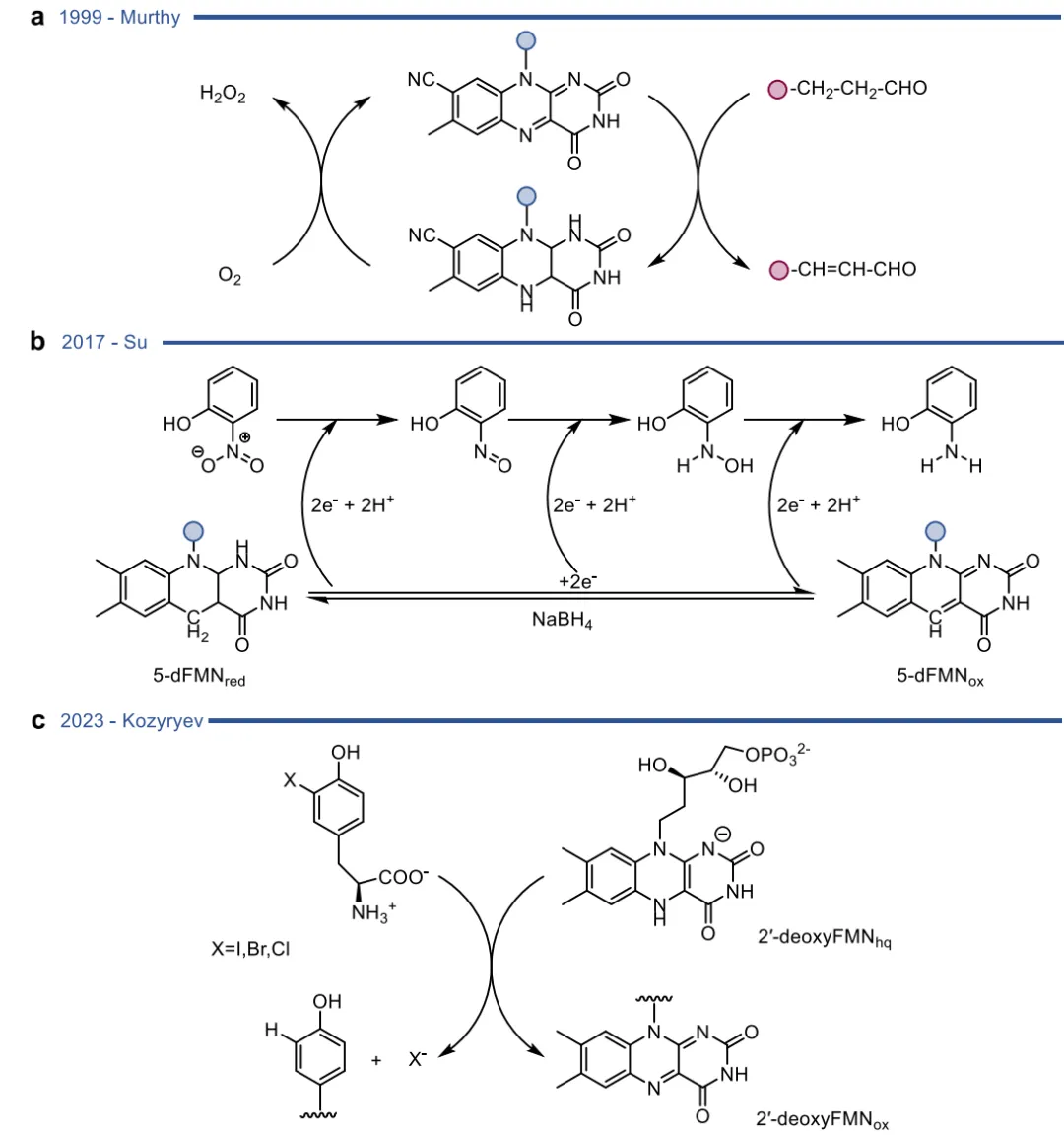
当酶的天然催化循环被完全绕过或彻底重构时,便进入了区别于自然机制的杂泛性领域 。这可以通过引入人工黄素辅因子来实现,例如将天然FMN替换为8-CN-FMN、缺乏单电子转移能力的5-脱氮黄素(5dFMN)或2'-脱氧FMN,从而在不改变蛋白质骨架的情况下彻底改变电子流动路径,将脱碘酶直接转化为氢负离子转移的硝基还原酶(图16) 。精准的蛋白质突变同样能实现深度的功能重写,如单一突变即可将PAMO从单加氧酶跨界转化为NADPH氧化酶,或使CHMO具备酮还原酶活性,以及让HMFO催化仲硫醇的氧化(图17) ;而针对老黄酶的改造,甚至能使其丧失原有的还原活性,转而催化Morita-Baylis-Hillman (MBH) 碳碳键偶联反应(图18) 。
图16. 利用修饰的人工黄素辅因子来调节酶功能
图17. 通过酶工程将PAMO突变体转化为NADPH氧化酶、将CHMO突变体转化为酮还原酶,以及HMFO突变体催化仲硫醇氧化
图18. OYE突变体催化的烯烃与醛之间的不对称Morita-Baylis-Hillman反应及其催化机制
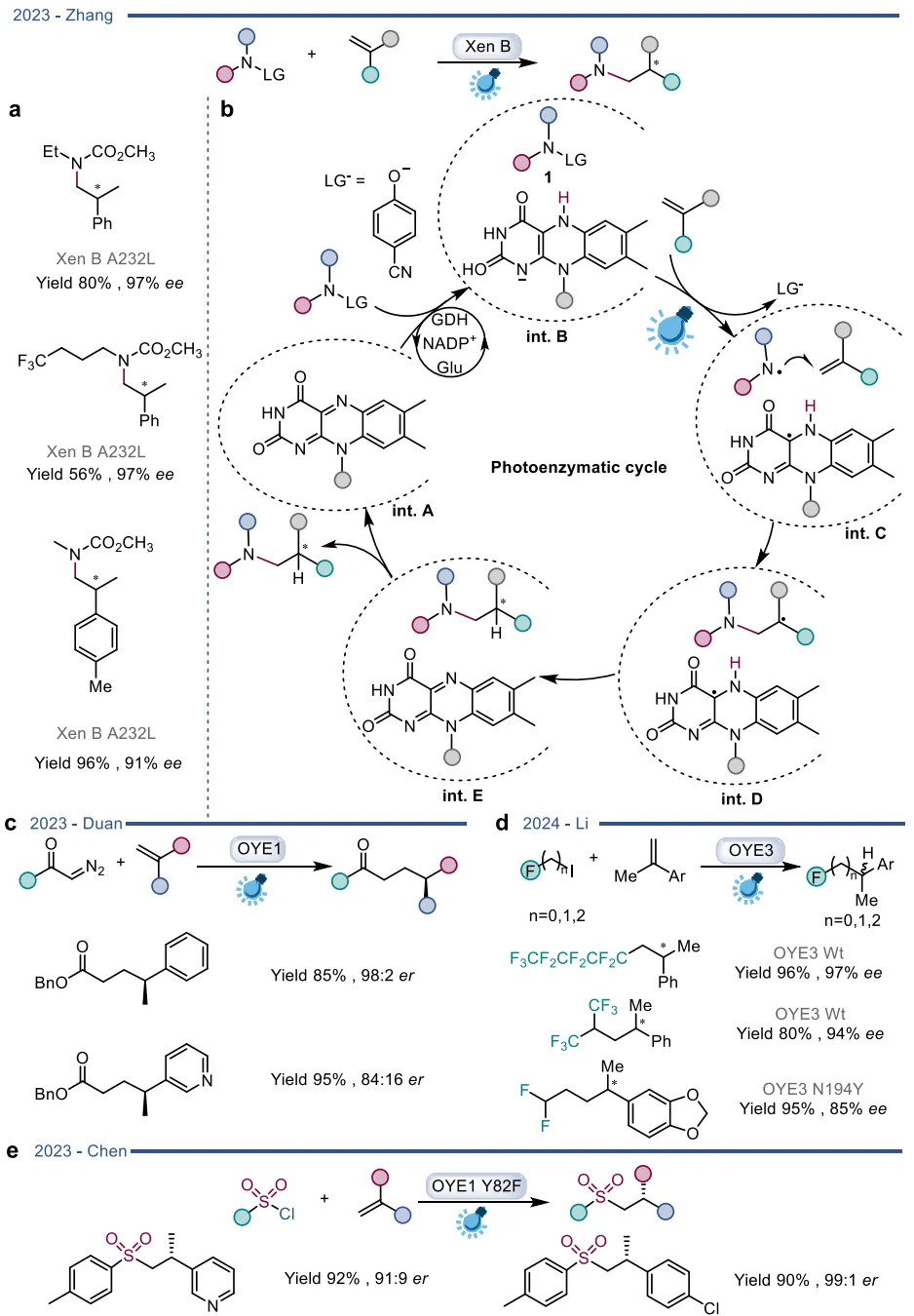
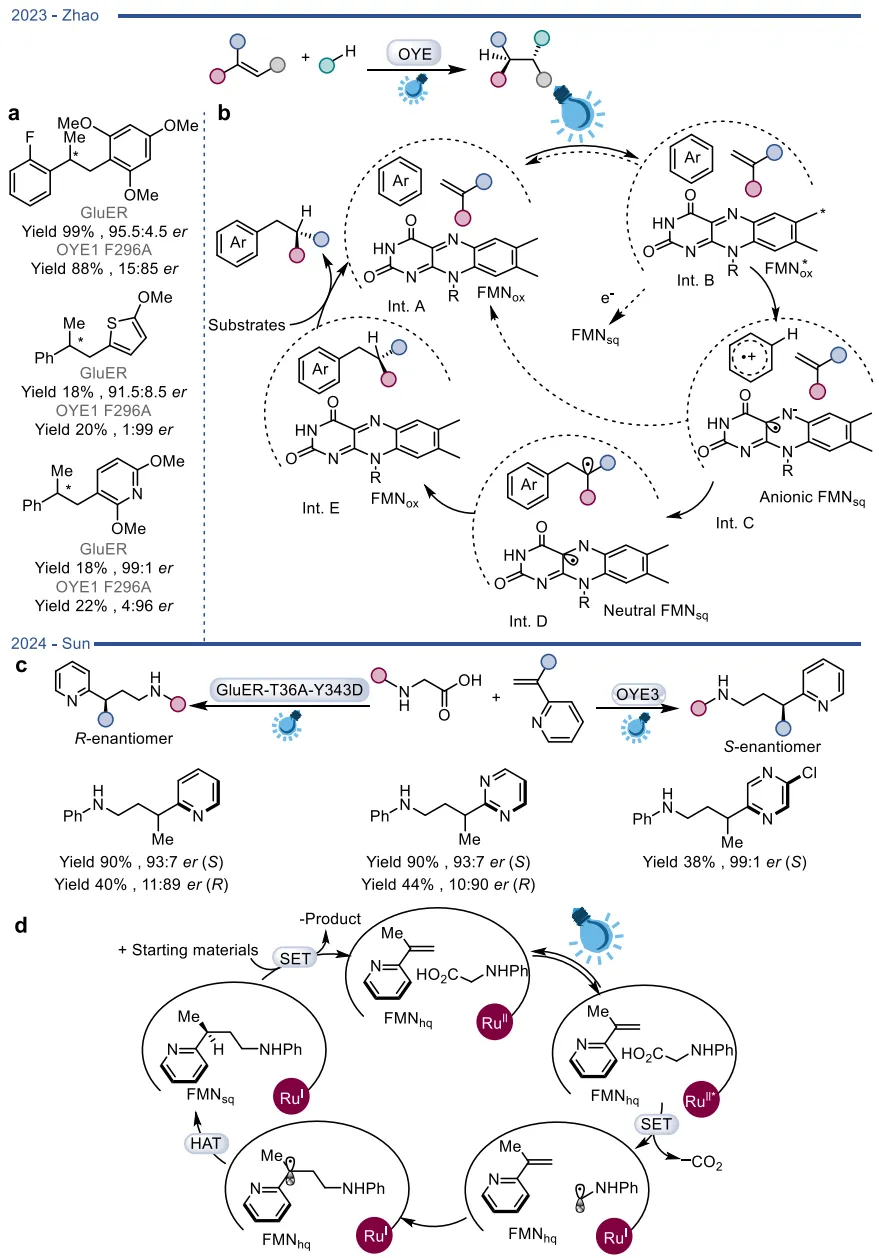
在彻底颠覆天然机制的探索中,酶促自由基反应尤其是光酶催化展现了极大的合成潜力 。在还原性自由基前体体系中,光激发的老黄酶能够通过电子转移引发自由基介导的高难度不对称碳氮键、碳碳键及碳硫键构建(图19) ;硝基还原酶与叶绿素等光催化剂结合,实现了协同的氧化还原级联(图20) 。在氧化性自由基前体体系中,光激发的黄素能够作为强氧化剂直接启动芳基化反应,或与钌配合物结合实现脱羧烷基化(图21) ;光活化的BVMOs则被用于催化未活化烯烃-胺底物的分子内氢胺化反应(图22) 。更为巧妙的是,利用还原态黄素的基态电子转移(Ground-state electron transfer)机制,烯还原酶可以在无光照条件下激发自由基中间体,驱动高对映选择性的自由基环化与硫醚偶联(图23),以及自由基三氟甲基化和脱卤反应(图24) 。黄素依赖型酶正从传统的氧化还原生物催化剂,快速演变为可高度定制化的多功能自由基及杂泛性催化平台 。
图19. 使用工程化OYE进行光酶催化
图20. 在光催化剂辅助的单电子转移与酶辅助的氢原子转移共同作用下,BaNTR1选择性还原硝基芳烃或硝基烷烃的反应及其机制
图21. 光酶催化的烯烃不对称自由基加氢芳基化反应、OYE催化的氧化脱羧烷基化反应,及其相应的催化机制
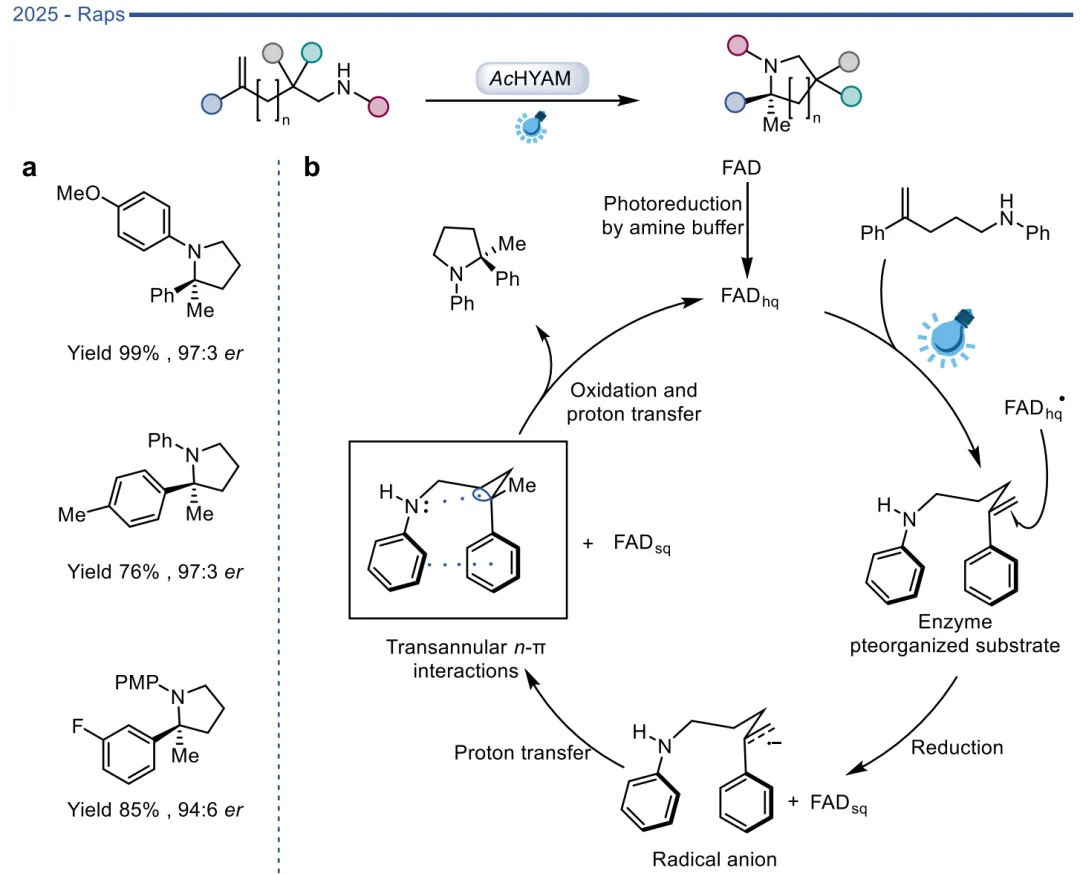
图22. AcHYAM介导的不对称氢胺化反应及其催化机制
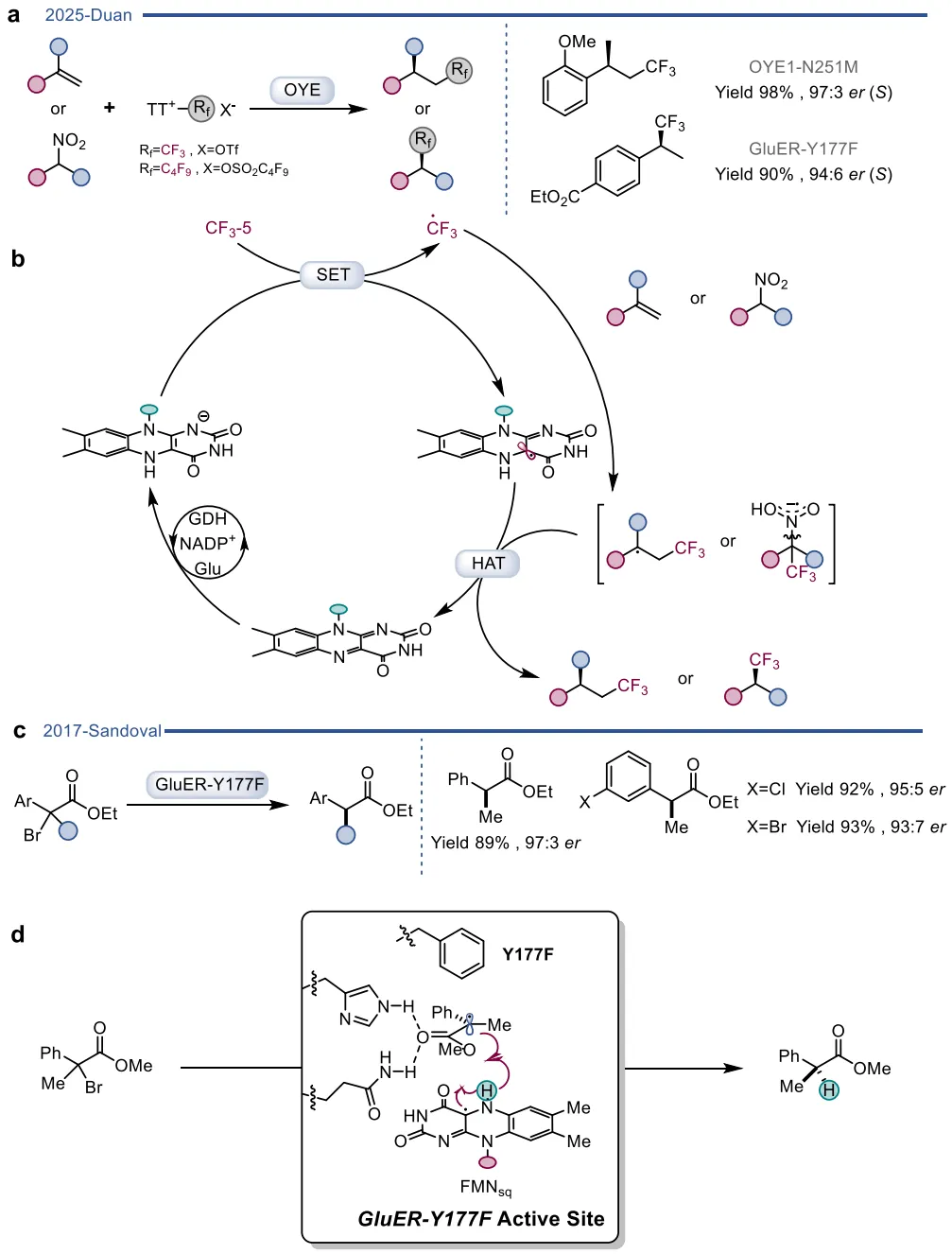
图23. ERED催化的不对称自由基反应
图24. 在无光照条件下通过基态电子转移机制实现的不对称自由基三氟甲基化和α-溴代酯不对称自由基脱卤反应及其相应的催化机制
过去十年间,得益于黄素辅因子的氧化还原灵活性及酶活性中心的可调性,黄素依赖型酶已从经典的氧化还原生物催化剂发展为具备高度适应性的多功能催化平台 。本综述明确区分了“基于自然机制的杂泛性”与“区别于自然机制的杂泛性”,指出辅因子替换、无氢供体催化、靶向蛋白质工程和光活化等系统扰动策略能有效解锁潜在或全新的反应范式,尤其是光酶催化在单电子自由基化学的立体控制方面展现出变革性潜力 。然而,该领域的工业化广泛应用仍面临三大关键挑战:首先是机制可预测性有限,因其广泛的反应性也会引发不需要的竞争途径,这需要结合高级光谱学、QM/MM-MD计算模拟和机器学习将经验观察转化为可靠的设计原则 ;其次是酶骨架多样性不足,亟需借助序列相似性网络分析、宏基因组挖掘、机器学习辅助工程以及合成生物学(如体内跨界光生物催化途径)来拓展可用框架并降低筛选成本 ;最后是光酶催化的工业放大规模受限,需要化学家、酶学家与工艺工程师跨学科协作,开发具备均匀光照、改进辅因子管理和高操作稳定性的标准化专用光生物反应器 。展望未来,通过将数据驱动的新酶挖掘(结合宏基因组学、机器学习与高通量定向进化)与机制驱动的反应设计(利用氧化还原态打破固有限制)相互融合互补,将高效推动黄素依赖型酶在全新非天然化学转化中的深度开发与工业应用 。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
