上海理工大学李贵生&南京理工大学&上海电力大学「国家杰青」李和兴,最新Nature子刊 | 晶格氧激活助力工业级海水电解!
- 2026-07-02 23:50:56

在本文中,作者展示了一种原位生长策略,将泡沫铁转化为硼掺杂的钴-铁羟基氧化物纳米片。硼掺入和无序晶格产生了丰富的氧空位,降低了水氧化的动力学能垒。该催化剂在碱性海水中仅需325 mV的过电位即可达到1.0 A cm-2的电流密度,并在600小时运行后性能衰减小于2%。
机理研究表明,氧空位激活了晶格氧氧化机制,绕过了传统吸附质演化机制的标度限制。无序结构增强了结构柔性以适应动态重构,从而减轻了活性位点的溶出。该策略可推广至其他过渡金属基羟基氧化物,实现大面积自支撑电极的制备。本工作为设计用于实际海水电解的耐久催化剂建立了一种通用策略。
利用间歇性可再生能源(如太阳能和风能)驱动盐水/海水电解,是生产清洁氢气最有前景且最可持续的方法之一,从而有助于应对能源危机和实现碳中和。与阴极上相对成熟的析氢反应技术相比,阳极上海水中的析氧反应面临着电极腐蚀、动力学迟缓以及不期望的氯析出反应等问题,这些阻碍了海水制绿氢的大规模实际应用。因此,设计合成低成本、高效、稳健的阳极催化剂是解决上述挑战的关键。
目前,贵金属基催化剂(如IrO2和RuO2)展现出最先进的OER性能;然而,它们的高昂价格和在地球上的有限储量成为限制其实际工业应用的瓶颈。此外,贵金属基催化剂在CER中也表现出高性能。海水中高浓度的氯离子(约0.5 M)会显著影响OER的法拉第效率,并转化为高腐蚀性的次氯酸盐物种,导致严重的电极腐蚀并损害催化耐久性。相关反应如下:
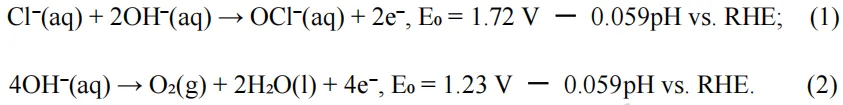
由地球丰富的过渡金属构成的过渡金属羟基氧化物是海水电解中贵金属基催化剂的替代品,在碱性条件下表现出高OER性能。先前的研究表明,MOOH的OER过程主要遵循两种反应机制:吸附质演化机制和晶格氧机制。其中,LOM路径涉及将晶格氧原子激活为直接参与O2形成的氧化还原活性中心,克服了AEM固有的动力学和热力学限制,从而表现出高OER性能。
然而,LOM路径中晶格氧原子的激活常常导致氧空位的形成和再填充,引起结构不稳定和催化性能下降。尽管有几个系统在500 mA∙cm-2下表现出显著的全水分解性能,但MOOH催化剂仍然存在本征活性有限和稳定性短(低于100小时)的问题。
考虑到碱性条件下竞争性CER的电位(0.89 V vs. RHE)比OER的电位(0.40 V vs. RHE)高出约480 mV,因此迫切需要开发能在低过电位(<480 mV)下达到工业级电流密度(>1.0 A cm-2)的OER催化剂,从而确保碱性海水环境中OER的高法拉第效率并最大限度地减少CER的发生。
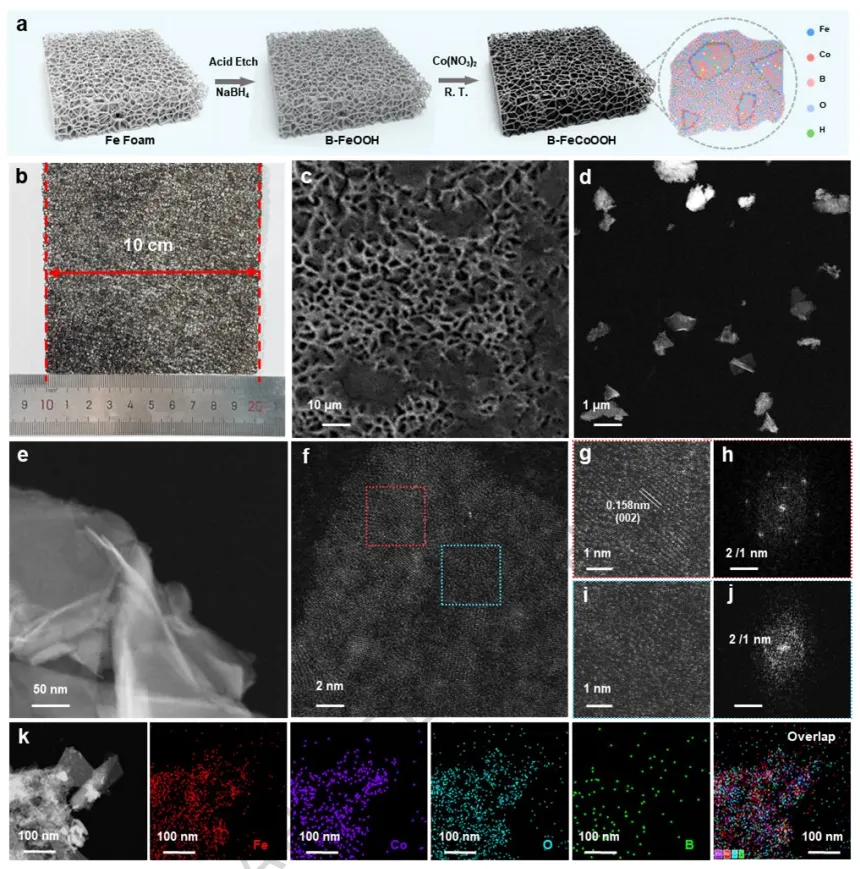
在本工作中,作者通过引入Co3+离子和稳定氧空位来优化FeOOH的能带结构。硼氢化物蚀刻的泡沫铁既作为结构模板又作为缺陷诱导剂,导致形成具有丰富结构缺陷的B-FeOOH。Co掺杂进一步调节了前驱体结构,促进了分子内电子再分布,并伴随着压缩晶格应变促进了高价Fe物种的生成。
在电氧化过程中,部分Fe的浸出诱导了无定形纳米片状羟基化金属氧化物的演变,这些氧化物整合到重构的FeCoOOH表面层中,从而最大限度地减少了活性物种和晶格氧的损失。Fe的浸出诱导了氧空位的形成,进一步驱动了电子再分布。
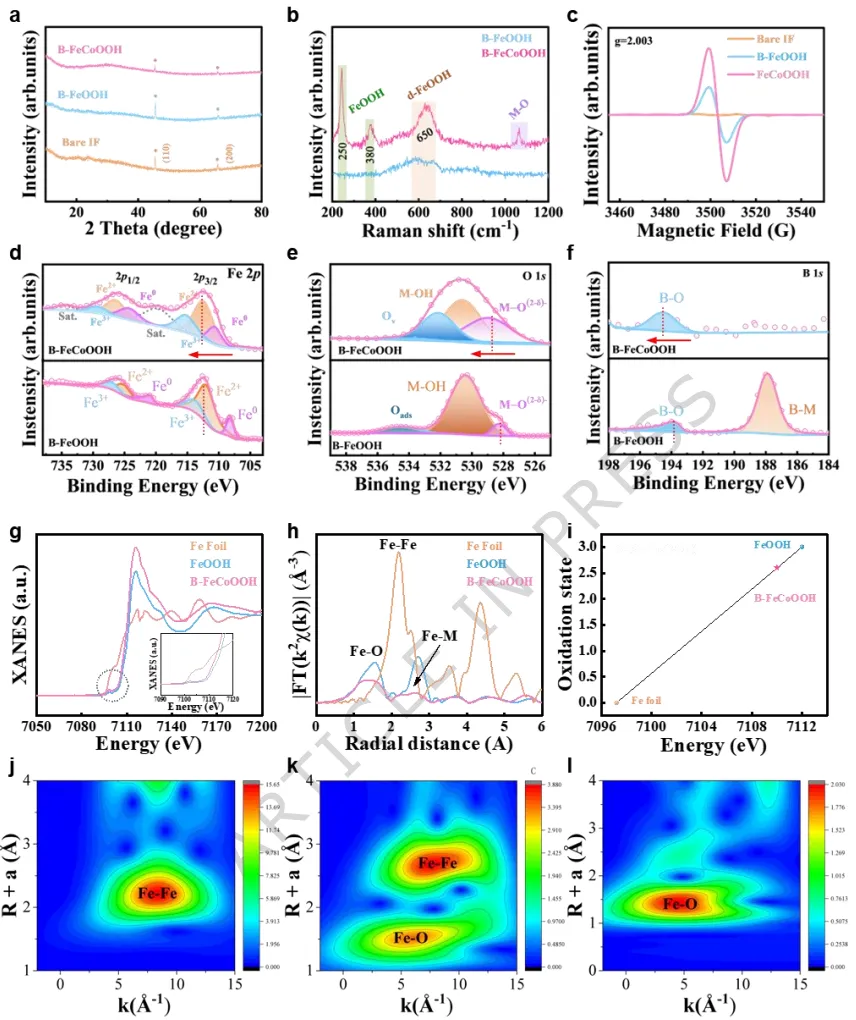
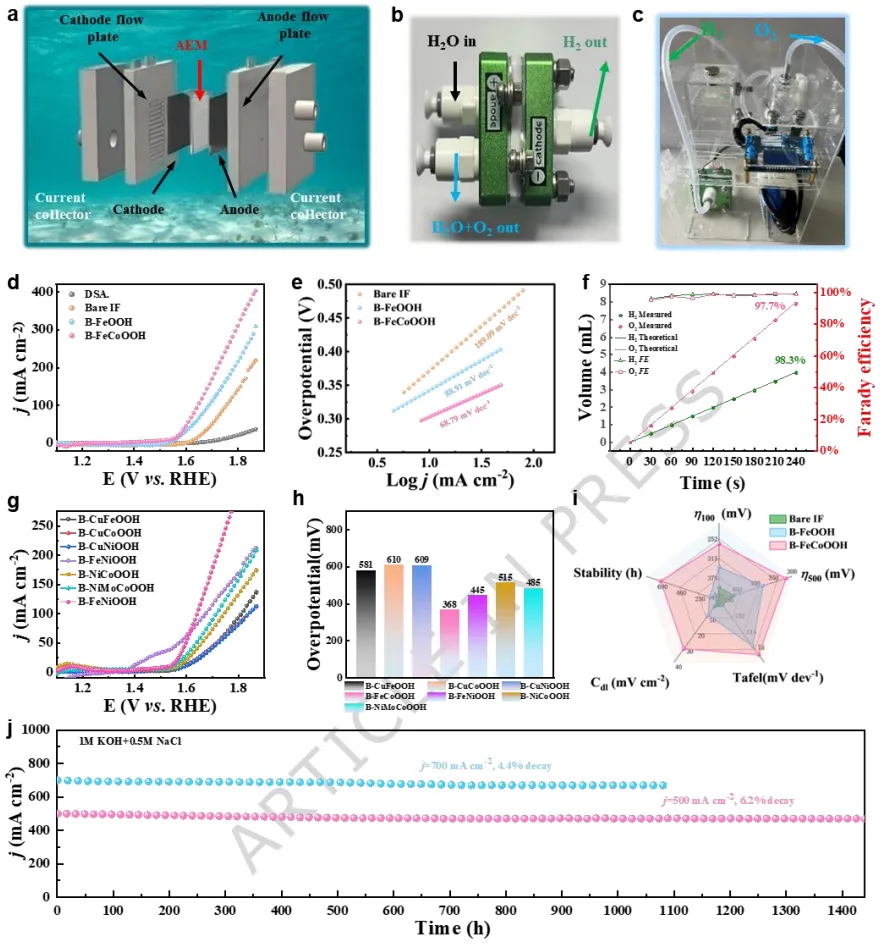
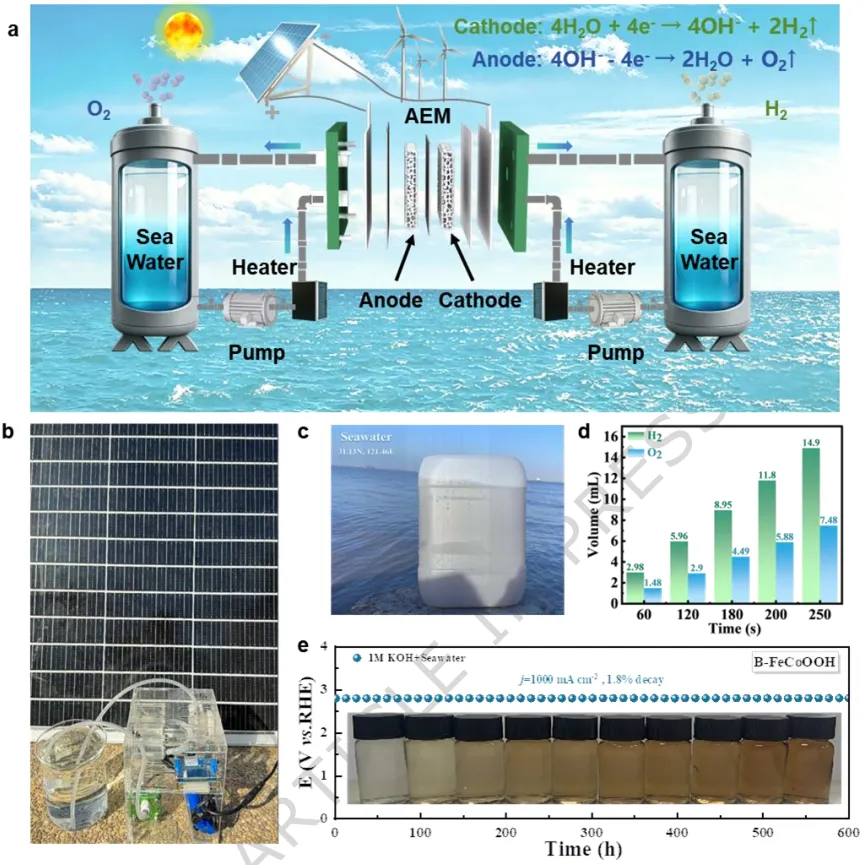
实验和理论分析揭示,阳极激活触发了金属和晶格氧位点的氧化还原过程,涉及氧空位的形成和补充。实验证实了晶格氧机制的存在,并通过决速步的转变降低了反应能垒。在碱性模拟海水中,B-FeCoOOH表现出高稳定性,在10和100 mA cm-2的电流密度下分别仅需186 mV和240 mV的过电位。
此外,该催化剂在0.5 A cm-2下稳定运行1440小时,在0.7 A cm-2下稳定运行1080小时,在工业级电流密度1.0 A cm-2下稳定运行600小时。值得注意的是,该系统在低过电位(<480 mV)下达到了1.6 A cm-2的工业级电流密度,确保了在碱性海水环境中的强Cl-耐腐蚀性。Fe浸出和稳定的氧空位的共存平衡了体系电荷,在OER循环中维持了氧空位的稳定性并防止了Fe3+的进一步浸出。
利用这一策略,成功制备了包含Co、Cu和Mo的耐腐蚀电极,实现了大规模、低成本的设计。这种方法不仅为构建羟基化金属氧化物提供了一种可推广的实验策略,而且通过精确调控氧空位和表面重构来调节活性位点形成,为增强OER性能的机制提供了有价值的见解。
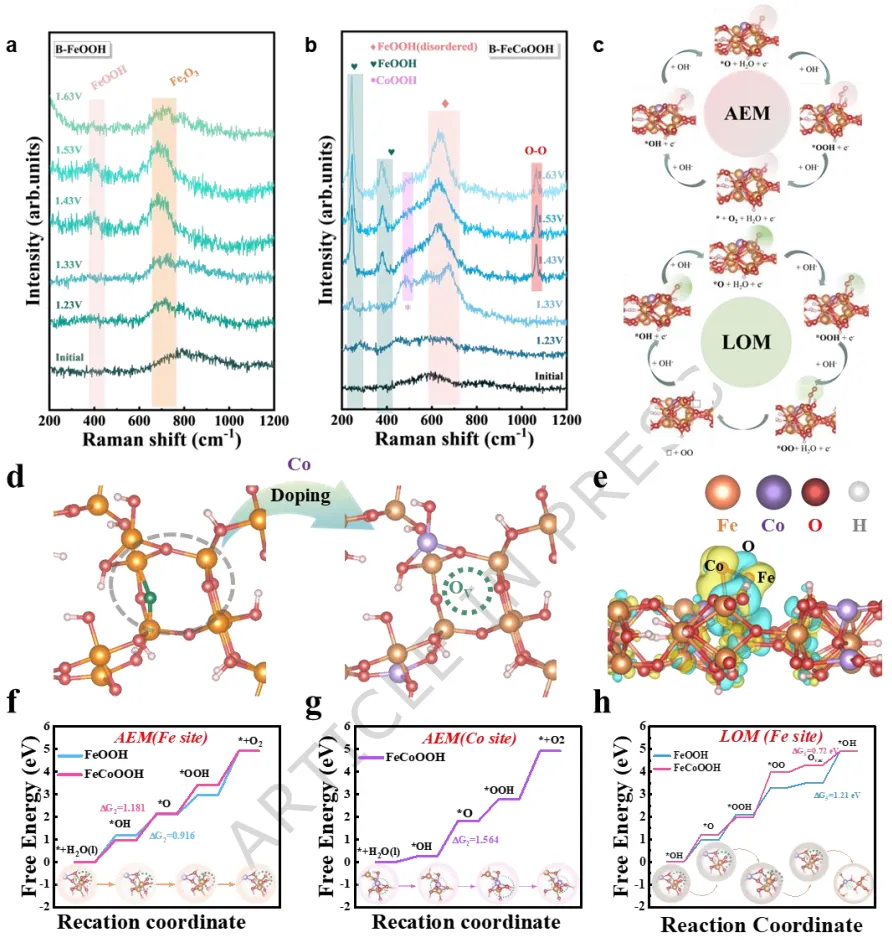
综上,该研究开发了一种原位生长策略,将泡沫铁转化为硼掺杂的钴-铁羟基氧化物纳米片催化剂(B-FeCoOOH)。硼掺入和无序晶格产生大量氧空位,激活了晶格氧氧化机制(LOM),绕过传统AEM路径的标度限制,显著降低了水氧化动力学能垒。
B-FeCoOOH在碱性模拟海水中仅需325 mV过电位即可达到1.0 A cm-2的工业级电流密度,在1.6 A cm-2下过电位低于480 mV,确保了对氯析出反应的高选择性。该催化剂在0.5 A cm-2下稳定运行1440小时,在0.7 A cm-2下稳定运行1080小时,在1.0 A cm-2下稳定运行600小时,性能衰减小于2%。
研究表明,Fe部分浸出诱导形成无定形羟基氧化物纳米屏障,与稳定存在的氧空位协同,防止了活性位点进一步溶出。该策略可推广至Fe、Ni、Cu等基底,以及Co、Cu、Mo等掺杂元素,实现了大面积自支撑电极的制备。结合太阳能光伏系统,该装置可稳定产氢产氧,为绿色氢能产业化提供了新思路。
Activating lattice oxygen in metal oxyhydroxides as durable electrodes for industrial-scale seawater splitting, Nature commumications, 2026,https://doi.org/10.1038/s41467-026-73894-4.

