ENO2⁺癌细胞通过MIF信号重塑免疫微环境,实验证实ENO2与MIF存在直接物理结合并精确定位互作界面,进而驱动M2巨噬细胞极化。这一机制揭示了ENO2介导免疫逃逸的新模式,提示阻断该互作或为逆转免疫抑制提供新策略(图4)。
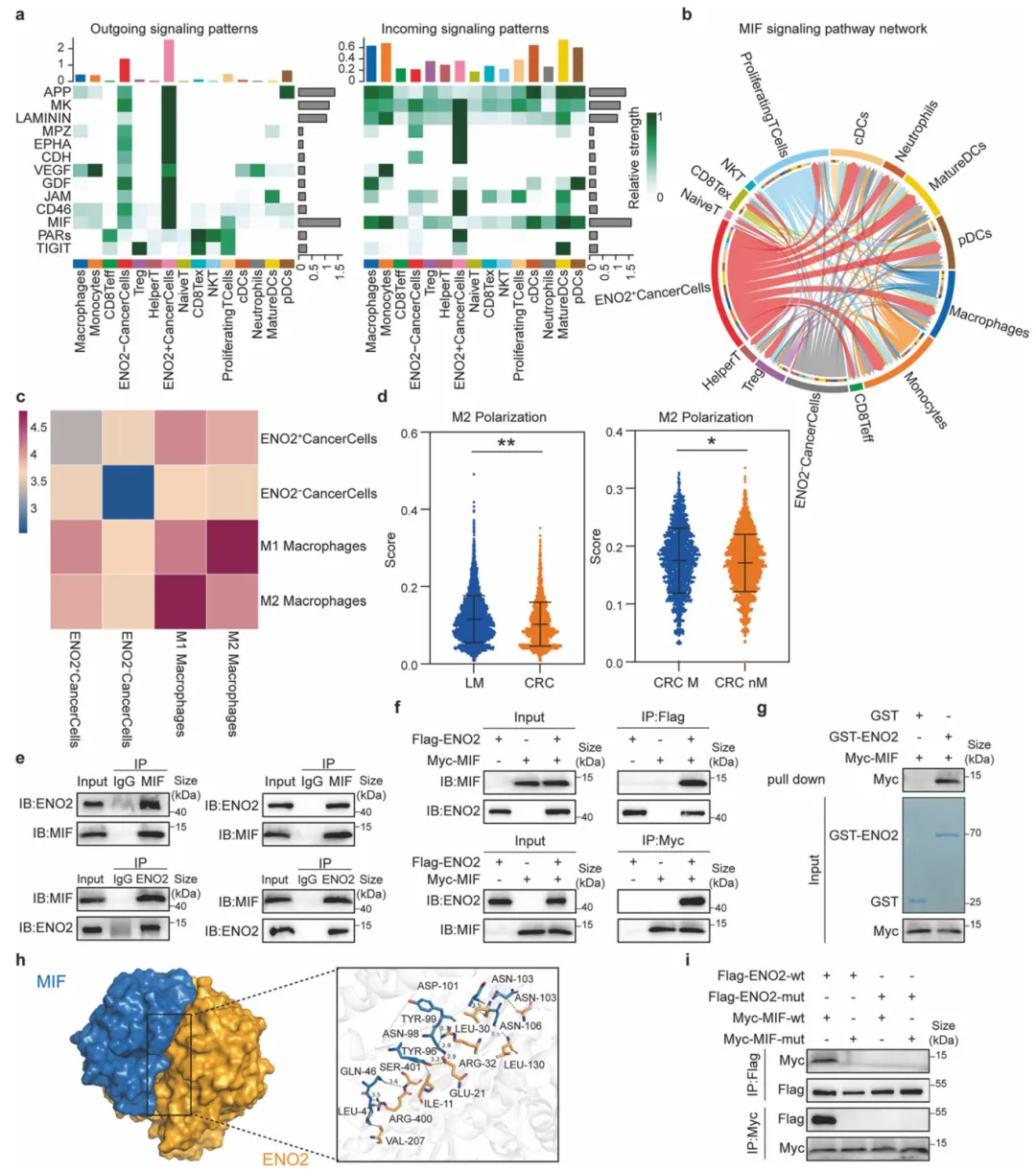
图4.ENO2通过与MIF直接相互作用,调控M2型巨噬细胞极化
利用CellChat进行细胞通讯分析,结合Co-IP与质谱鉴定互作蛋白,采用GST pull-down验证直接结合,并借助分子对接与定点突变技术精确定位蛋白互作的关键结构域。
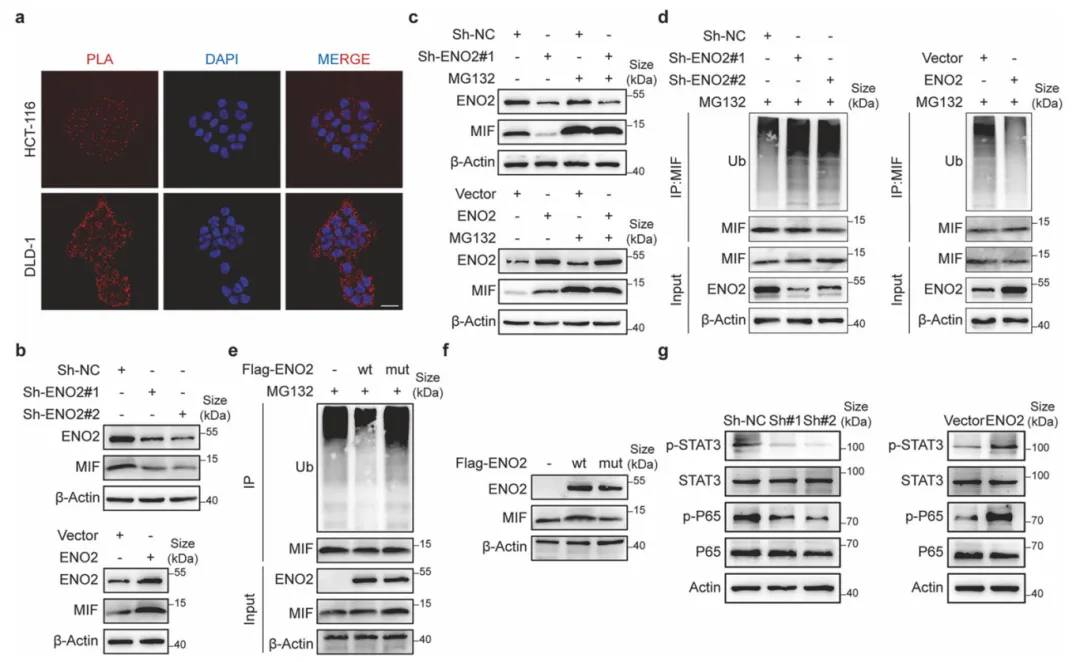
ENO2通过直接结合MIF,阻断CHIP介导的多聚泛素化修饰,拮抗蛋白酶体降解,从而显著提升MIF蛋白稳定性。该机制依赖完整的结合界面,并持续激活下游STAT3与NF-κB信号轴,构成了驱动结直肠癌转移的关键分子开关(图5)。
图5.ENO2通过抑制泛素介导的降解来稳定MIF蛋白
采用PLA技术验证蛋白原位互作,利用MG132处理区分转录与翻译后调控,结合Co-IP及泛素化检测,通过回补野生型与降解缺陷型MIF验证其对下游信号通路的特异性调控。
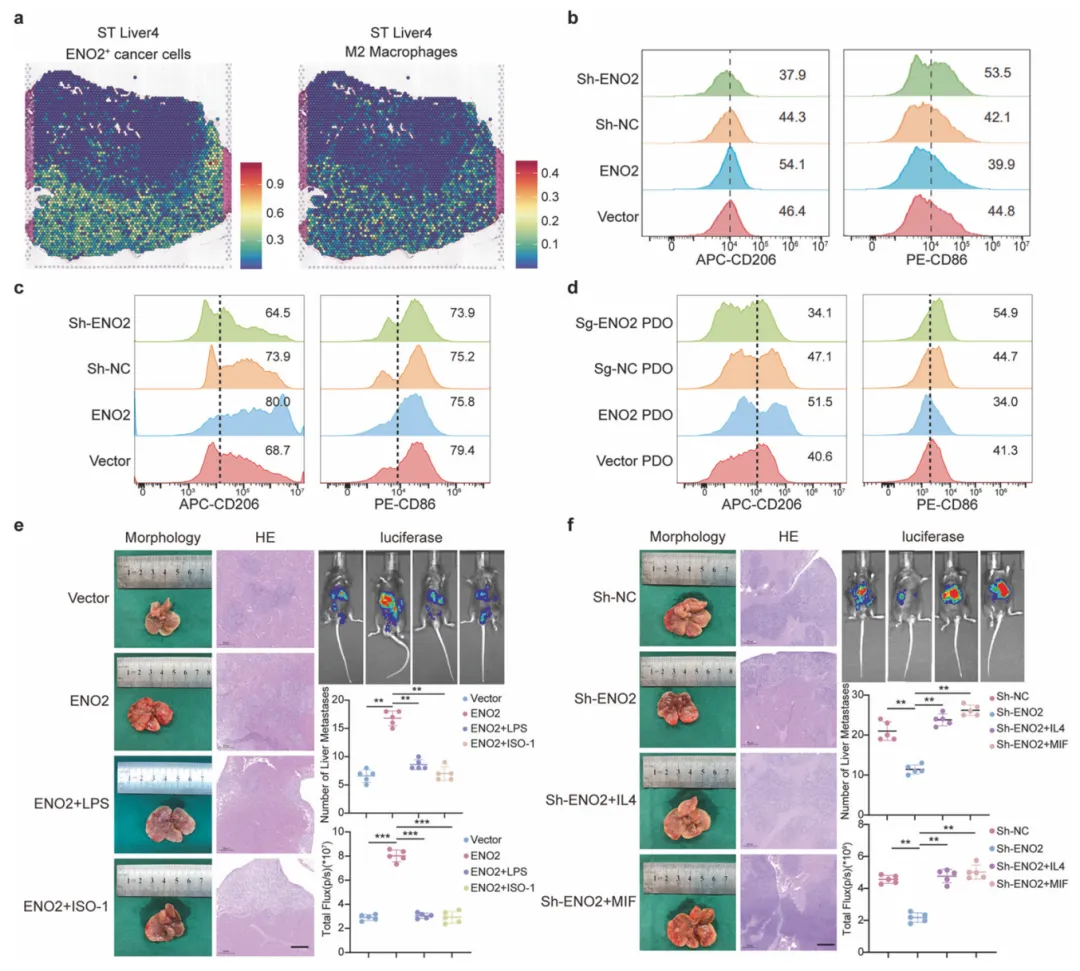
ENO2⁺癌细胞借MIF诱导M2巨噬细胞极化,驱动肝转移。空间共定位与体内外实验佐证该轴必要性,抑制MIF或M2极化可阻断转移。但模型未能完全模拟临床免疫微环境,且忽略了其他免疫细胞亚群的潜在调控作用(图6)。
图6.ENO2诱导M2型巨噬细胞极化以促进肝转移
结合空间转录组(10x Visium)定位细胞邻域,利用Transwell或流式细胞术检测共培养后巨噬细胞标志物变化,并通过小鼠脾注射模型验证ENO2-MIF轴对肝转移的调控功能。
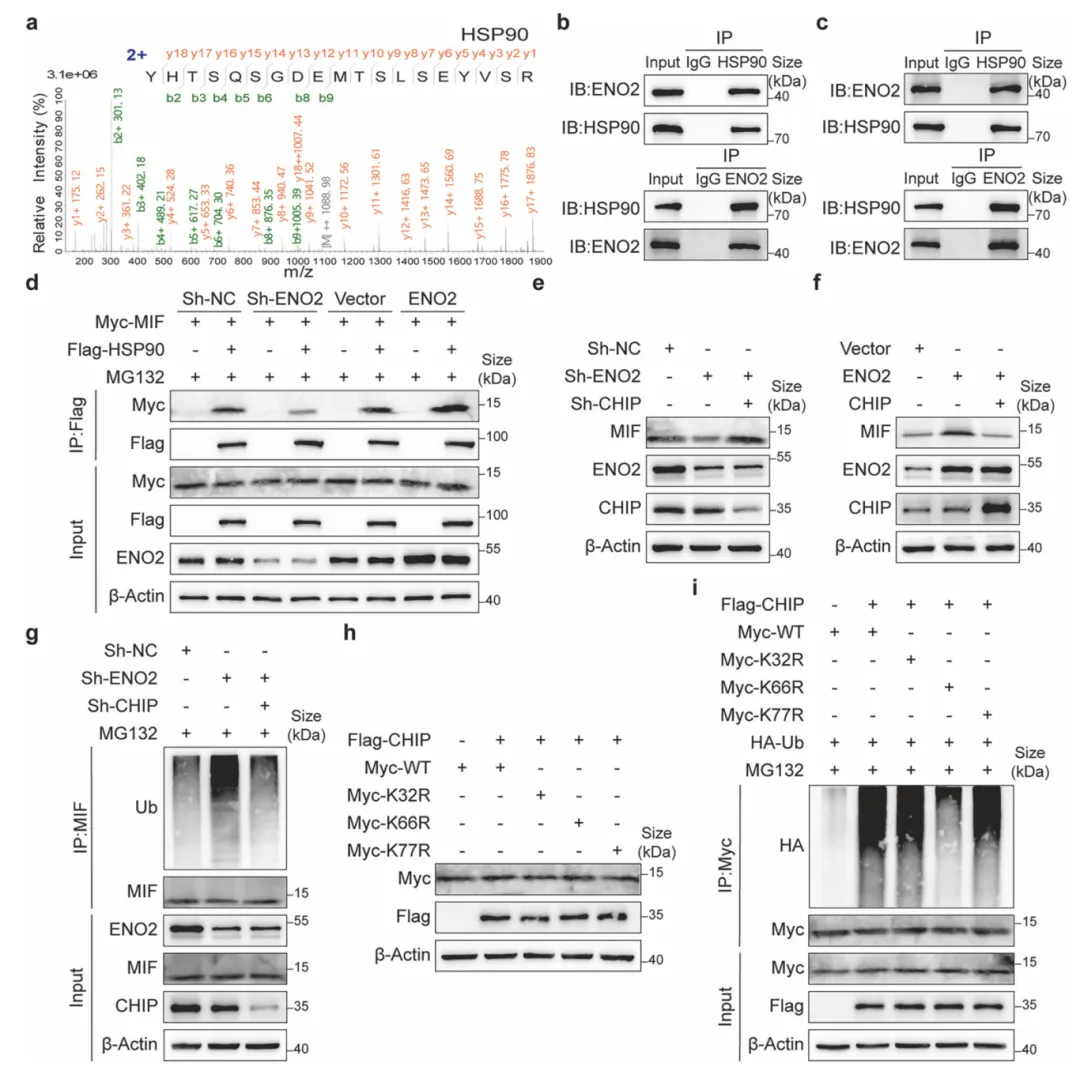
ENO2作为支架招募HSP90,通过空间位阻阻断E3连接酶CHIP对MIF的K66位点泛素化修饰,从而拮抗其降解。回复实验证实,耗竭CHIP可逆转ENO2缺失导致的MIF失稳,完善了ENO2-HSP90-CHIP调控轴(图7)。
图7.ENO2招募HSP90以拮抗CHIP介导的MIF泛素化与降解
采用Co-IP联合质谱鉴定互作蛋白,构建MIF赖氨酸位点突变体(K66R),利用泛素化检测与CHIP敲低回复实验,验证三元复合物对底物蛋白稳定性的调控机制。
八、靶向ENO2-MIF互作的小分子抑制剂阻断肝转移
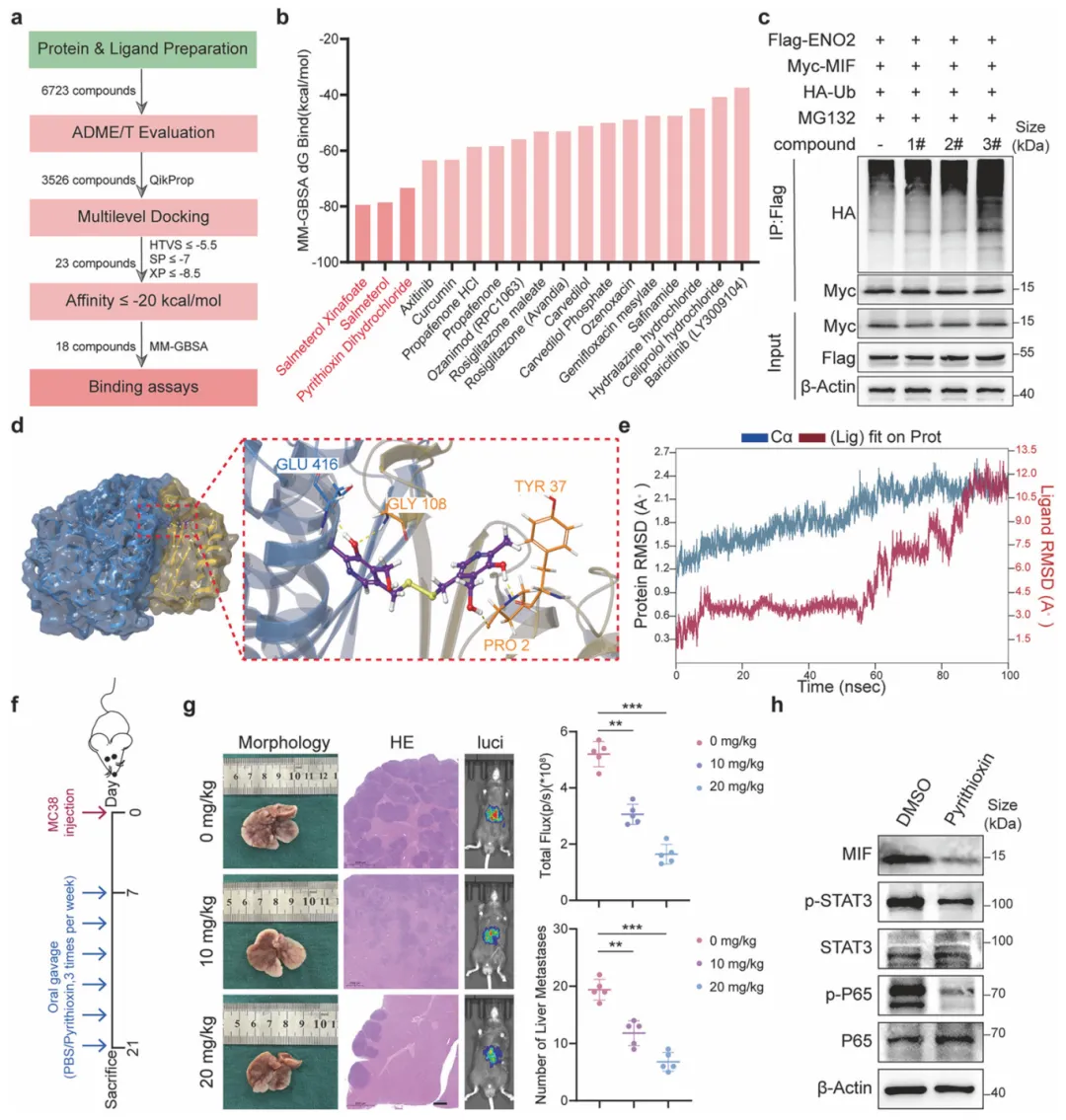
虚拟筛选鉴定出ENO2-MIF互作抑制剂吡硫醇,其通过恢复MIF泛素化降解、阻断STAT3/NF-κB信号,显著抑制小鼠肝转移并重塑免疫微环境。这为靶向蛋白互作界面干预结直肠癌转移提供了概念验证(图8)。
图8.pyrithioxin作为ENO2-MIF相互作用抑制剂的鉴定及其体内抗转移效果验证






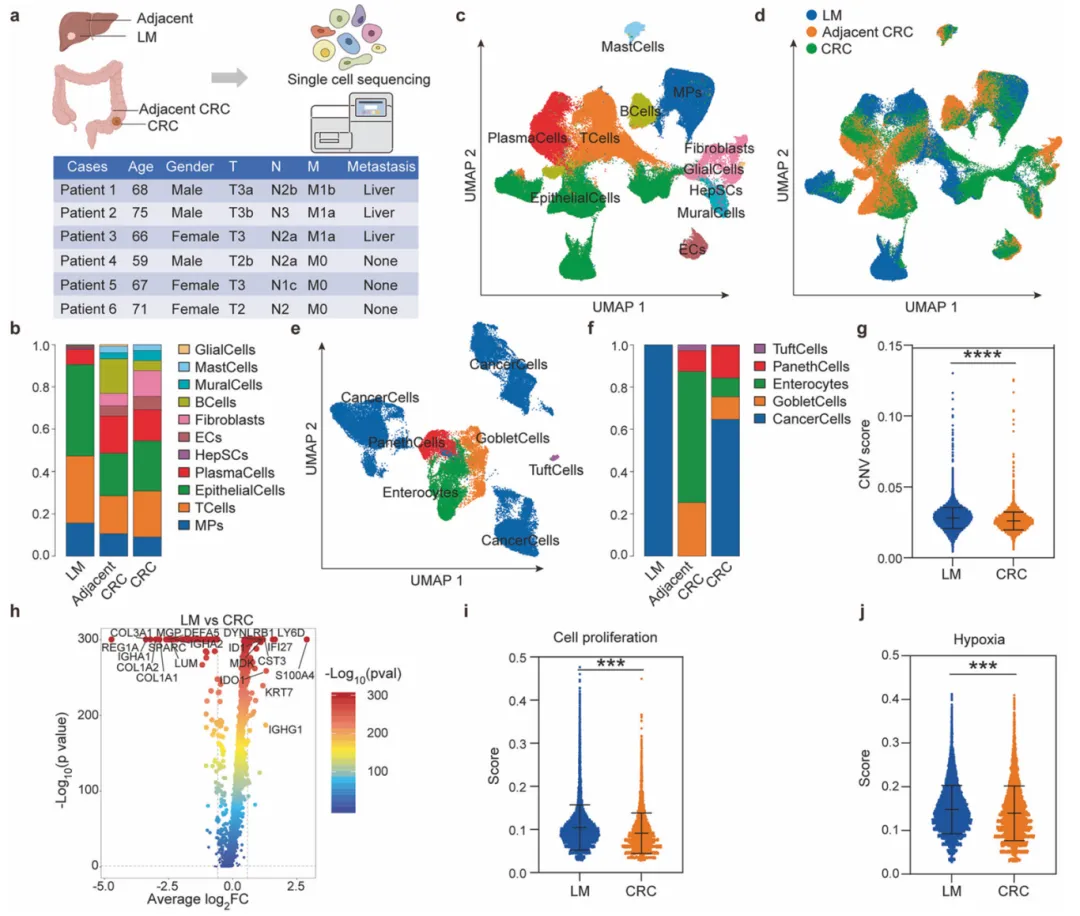

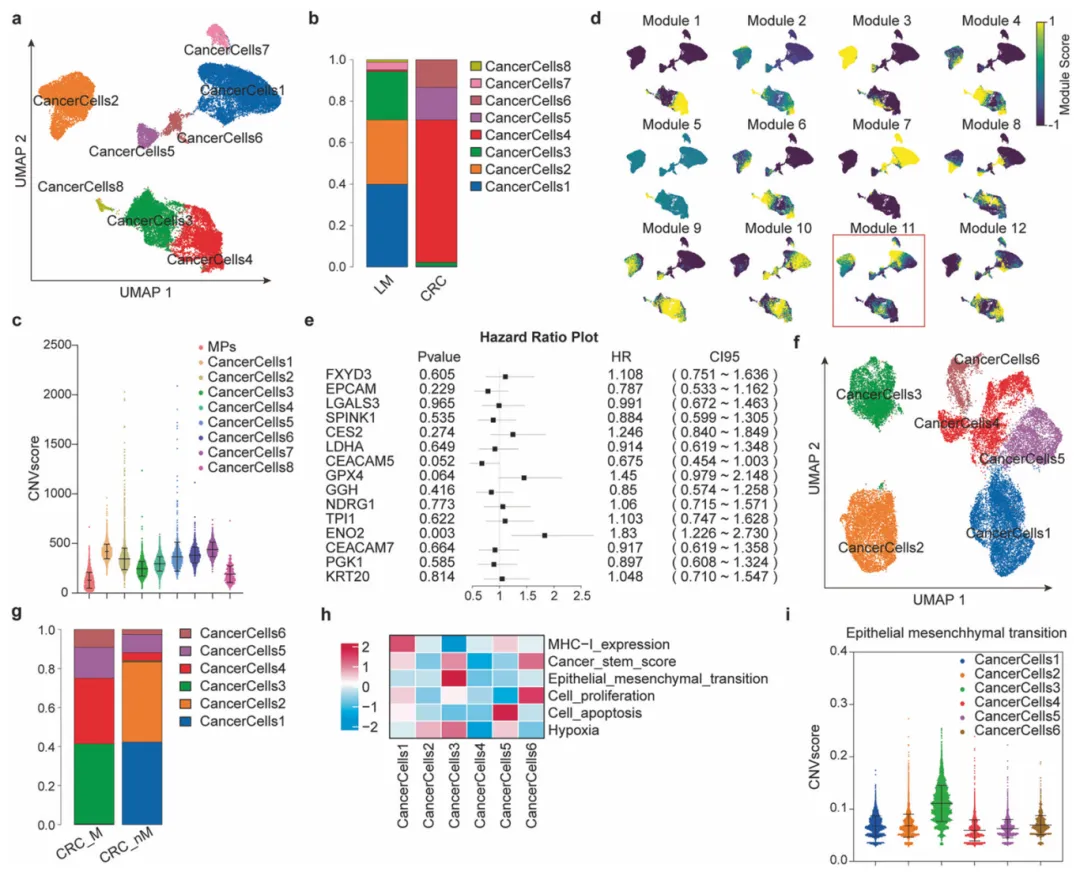

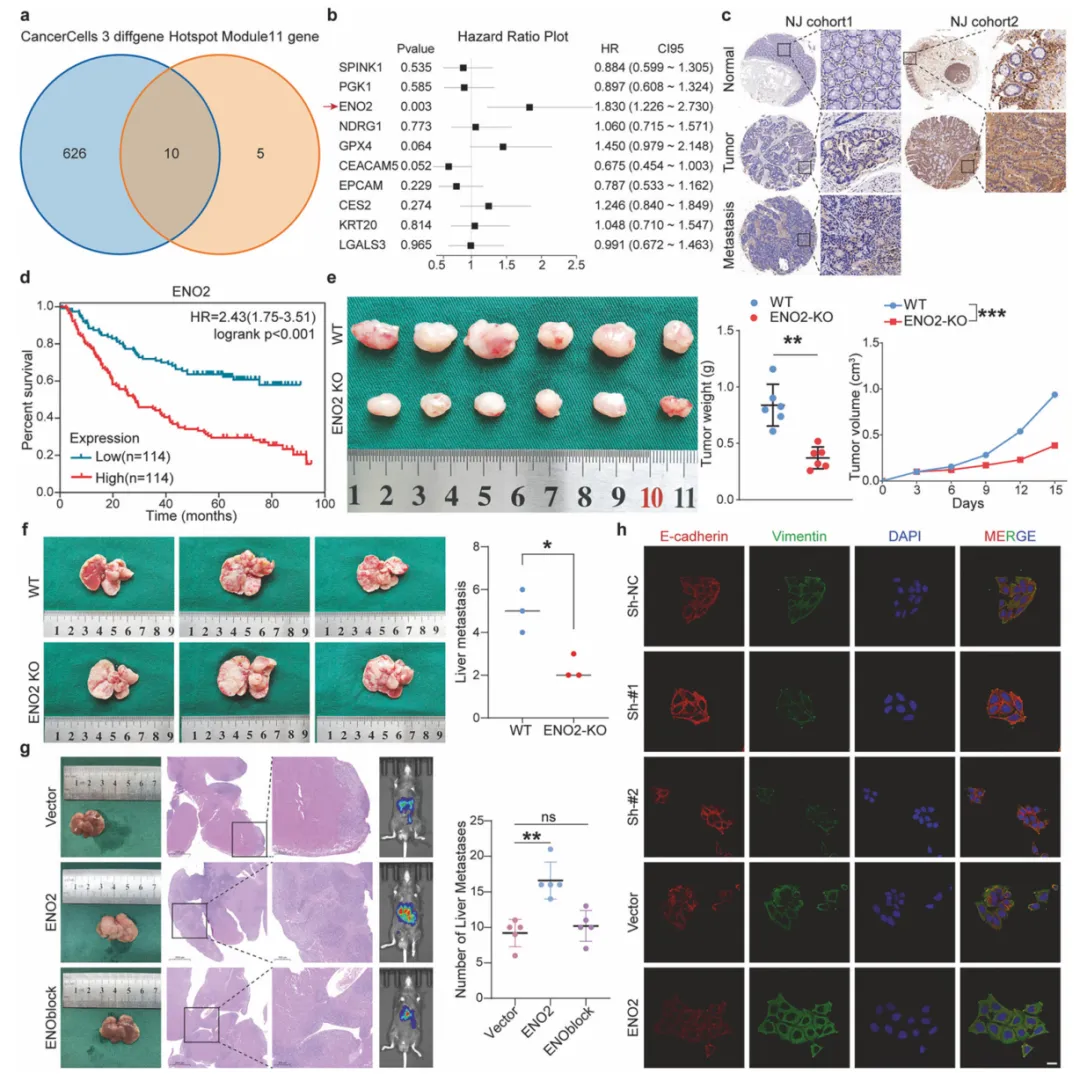








 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
