【JACS】南京工业大学郑旦庆课题组:N-亚磺酰基邻苯二甲酰亚胺:模块化亚磺酰自由基源及其催化的烯烃双官能团化与自由基交叉偶联反应
- 2026-06-28 08:01:57

背景介绍
磺氧化物广泛存在于临床药物(如奥美拉唑)及天然产物中,传统合成方法依赖硫化物氧化或磺酸盐与有机金属试剂偶联,存在过度氧化或需预活化底质的局限。尽管硫中心自由基构建含硫骨架具有条件温和的优势,但相较于硫基(thiyl)和磺酰基(sulfonyl)自由基,亚磺酰自由基(RSO•)因易可逆加成至π体系及均二聚生成硫代磺酸酯而难以捉摸;此前Bi课题组开发的亚磺酰砜(sulfinyl sulfone)虽可同时释放亚磺酰与磺酰自由基,但共生成的活泼磺酰自由基干扰了持久自由基效应(PRE)所需的理想动力学平衡,限制其在多组分反应中的应用。因此,开发能"清洁"、单一释放持久性亚磺酰自由基且稳定性好的模块化前体是该领域的关键瓶颈。
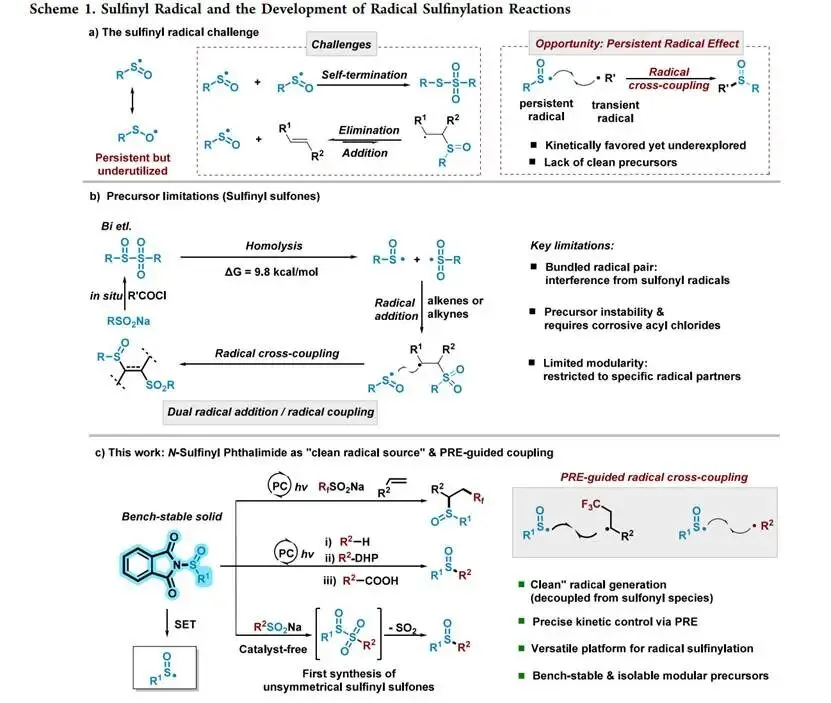
研究思路
受N-羟基邻苯二甲酰亚胺酯(RAE)通过单电子还原发生N—O均裂释放烷基自由基的启发,作者设计将亚磺酰基连接于邻苯二甲酰亚胺氮上,构建N-亚磺酰基邻苯二甲酰亚胺(N-sulfinyl phthalimide)。该分子在光催化剂还原态的单电子转移(SET)下发生N—S键介解断裂(mesolytic fragmentation),定量释放出目标亚磺酰自由基及惰性的邻苯二甲酰亚胺阴离子——后者不参与后续自由基链传递,从而在热力学和动力学上完美模拟PRE要求的"持久自由基+瞬态自由基"二元体系,规避了以往共生成副自由基的问题,并可从廉价易得的亚磺酸钠盐简便制备得到空气稳定的结晶固体。
研究内容
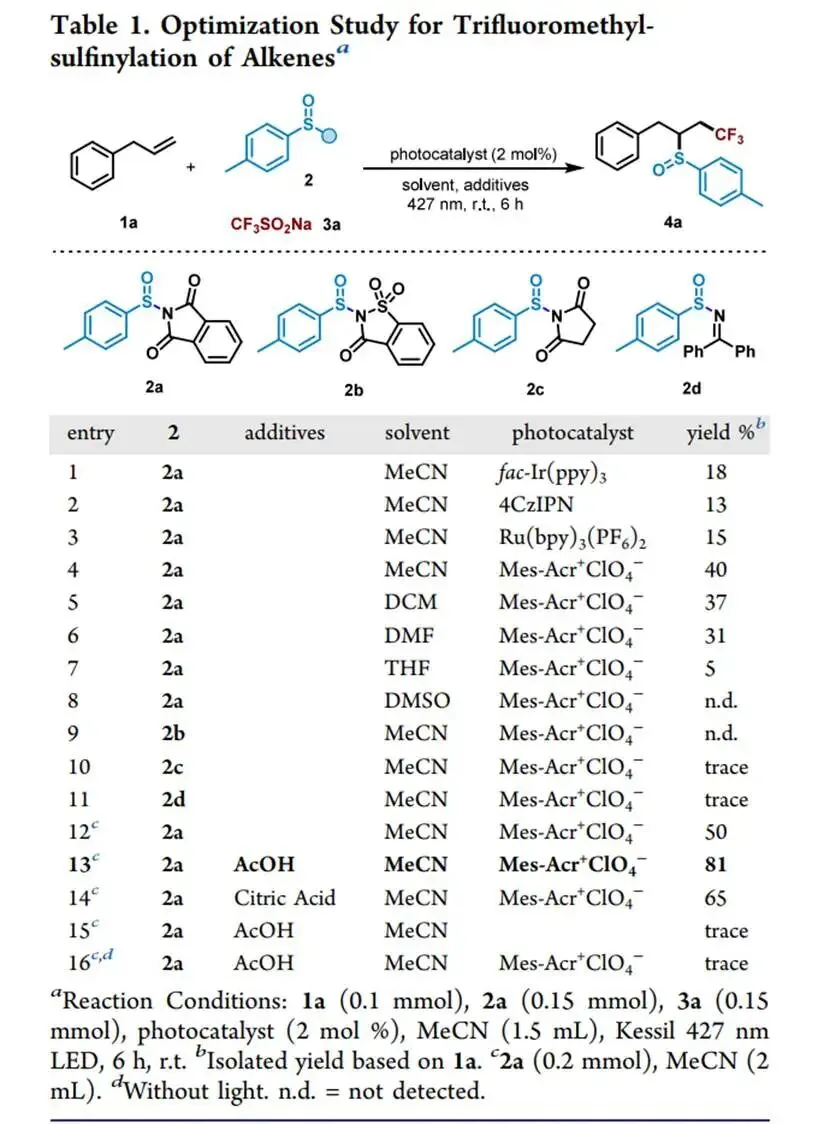
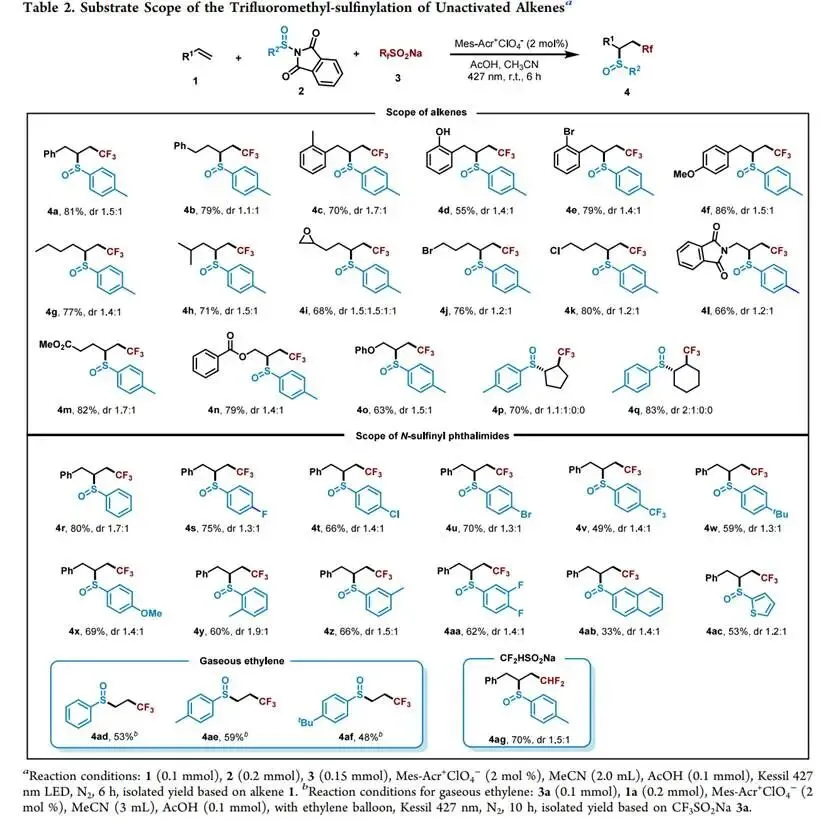
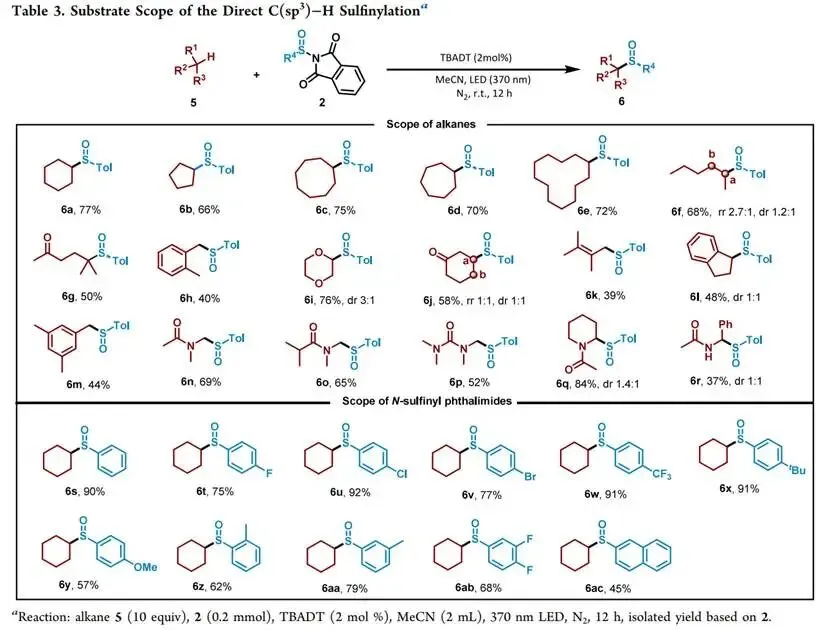
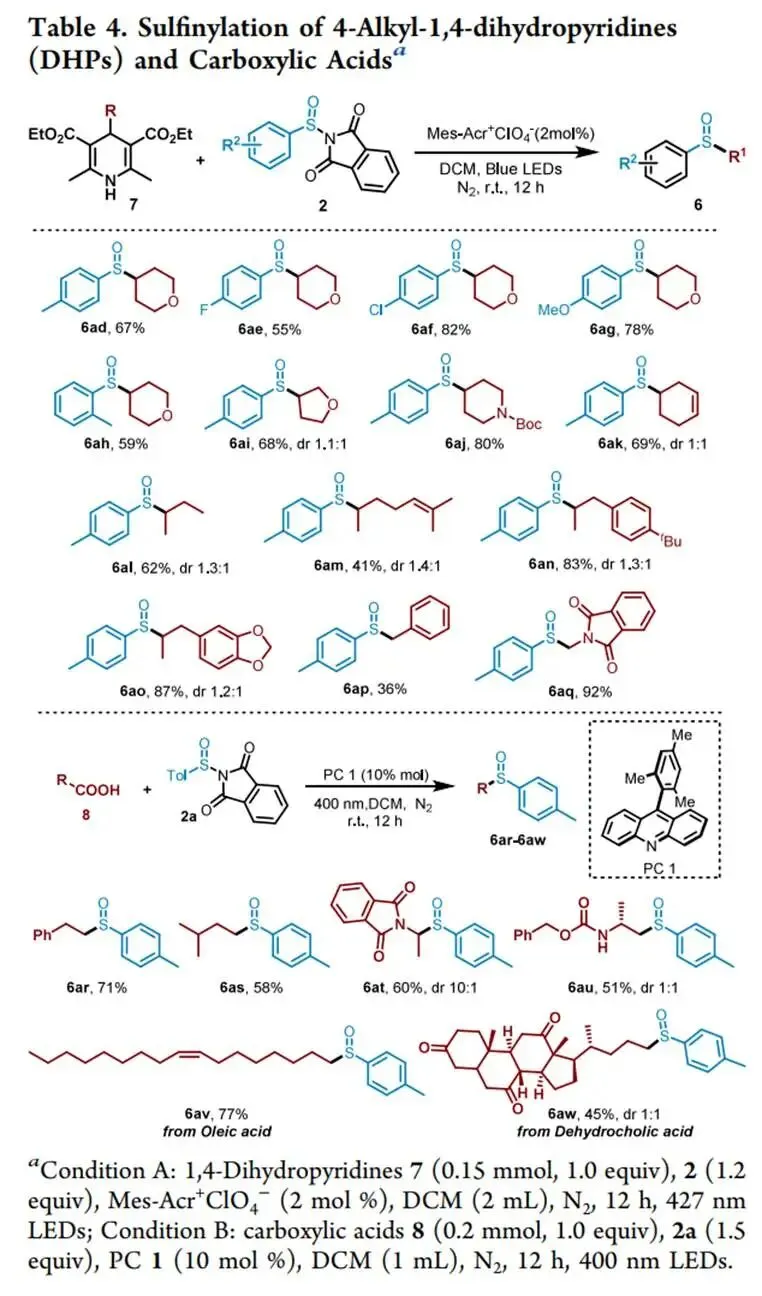
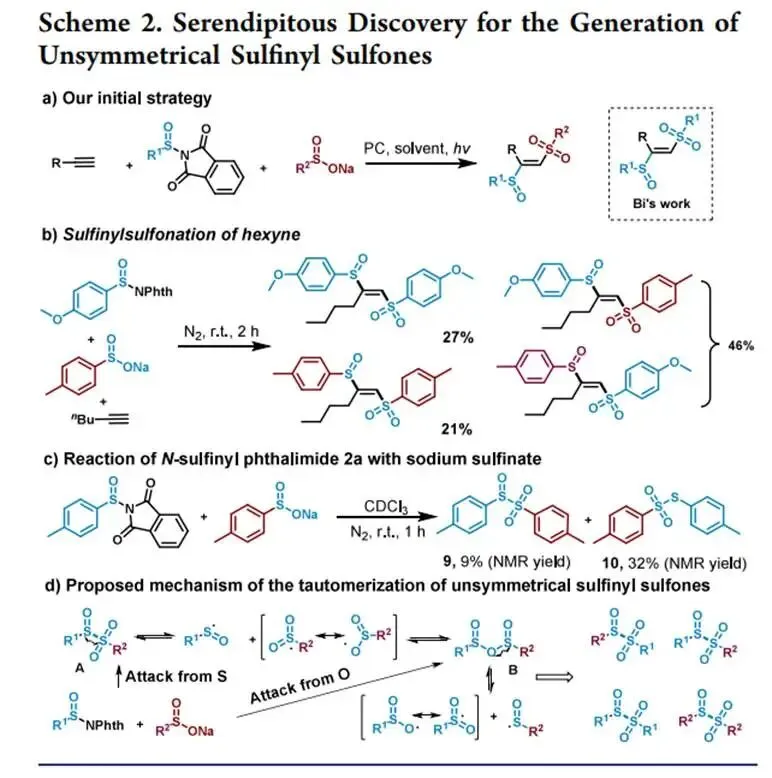
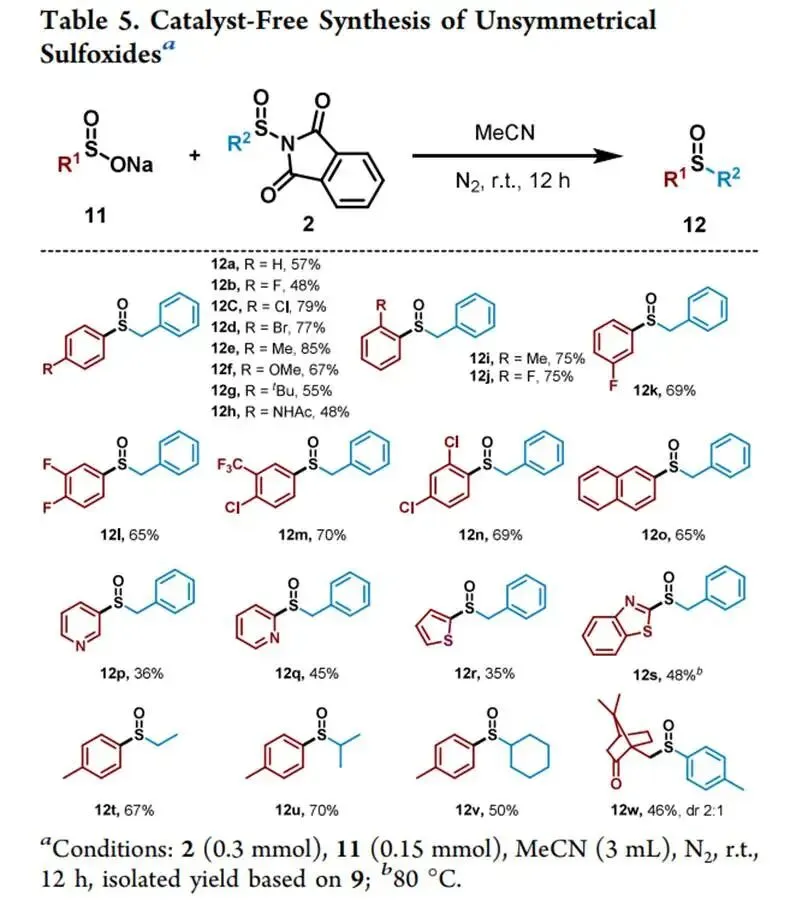
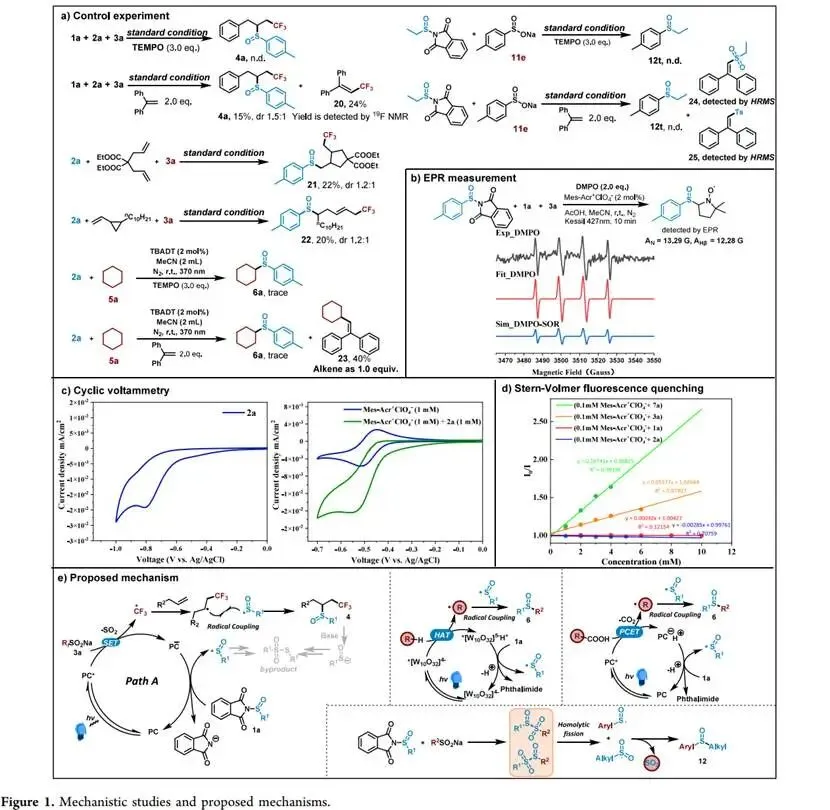
在427 nm蓝光/Mes-Acr⁺ClO₄⁻催化下,以烯丙苯、N-对甲苯亚磺酰基邻苯二甲酰亚胺和CF₃SO₂Na为模板,优化加乙酸抑制碱促分解后,实现了未活化烯烃高化学选择性1,2-三氟甲基-亚磺酰化(产率最高85%),兼容环内烯(反式选择性)及多种敏感官能团,并可扩至乙烯气体双官能团化;切换自由基源可实现C(sp³)—H键(十钨酸铵HAT活化,烷烃仅需10当量)、4-烷基-1,4-二氢吡啶及羧酸(脱羧)与亚磺酰自由基的PRE导向交叉偶联,广谱获取α-位取代磺氧化物及复杂天然产物衍生磺氧化物。意外发现N-亚磺酰基邻苯二甲酰亚胺与磺酸钠盐在室温无催化剂下经亚磺酰砜中间体、S—S键均裂/SO₂挤出/自由基重组,可高效合成不对称磺氧化物。机理研究通过TEMPO猝灭、自由基钟、EPR捕获DMPO—RSO•加合物及循环伏安法证实:光催化循环中激发态光催化剂氧化磺酸盐产生·CF₃,还原态PC⁻使N-亚磺酰基邻苯二甲酰亚胺发生SET诱导N—S断裂放出持久性RSO•,二者交叉偶联得目标产物。
总结
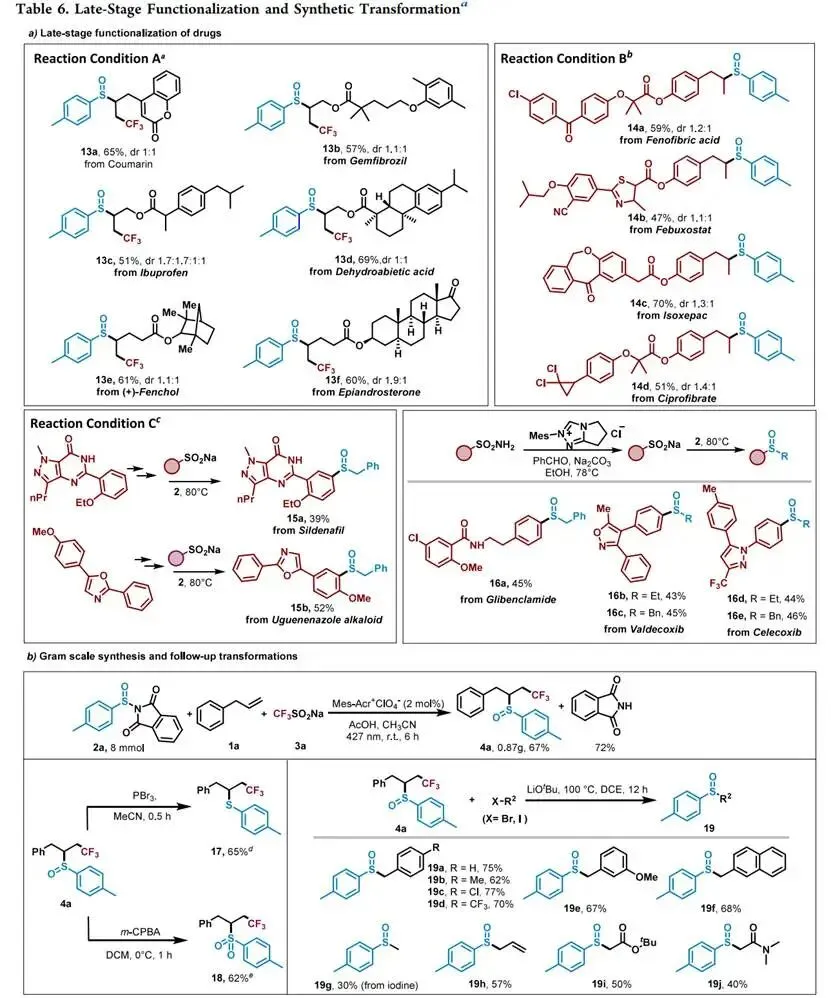
本研究首创N-亚磺酰基邻苯二甲酰亚胺作为空气稳定、模块化的"清洁"亚磺酰自由基前体,借助PRE动力学控制实现了未活化烯烃的三氟甲基硫亚磺酰化及多样化烷基自由基—亚磺酰基交叉偶联;意外发现的磺酸盐与该类试剂无催化剂室温偶联为不对称磺氧化物合成提供新途径,并首次表征了不稳定的不对称亚磺酰砜中间体及其互变异构行为。该方法操作简便、官能团耐受性好、可克级放大并回收副产物邻苯二甲酰亚胺,成功应用于药物分子的后期磺酰化修饰,为磺氧化物合成及硫自由基化学建立了通用平台。
文献详情
- 作者:Changmei Zhang, Yuxuan He, Heping Tan, Yang He, Danqing Zheng
- 题目:N-Sulfinyl Phthalimides as Modular Sulfinyl Radical Precursors: PRE-Guided Chemoselective Alkene Difunctionalization and Radical Cross-Coupling
- 期刊:Journal of the American Chemical Society (JACS)
- DOI:https://doi.org/10.1021/jacs.6c05222
(所有图片来自网络,如有侵权联系删除)
看往期精彩文章 ↓
【JACS】NUS赵宇/天大福州学院杨彬淼团队:钯催化不对称环加成构建含手性C-N轴手性及中心手性的苯并稠合中环杂环及骨架重排研究
【Nat. Chem.】天津大学福州国际联合学院赵宇/杨彬淼团队联合蓝宇实现中环合成新突破
如果我们的文章对你有帮助, 点击左下角“+关注 ”!这样就不会错过每次的推送啦!
右下点赞、点分享、点推荐,就是最好的支持

