第一作者:李晓蕊、陶浩兰
通讯作者:黄宏文、高磊
通讯单位:湖南大学、南京大学
论文DOI:10.1002/anie.7341900
在铂基氧还原反应(ORR)催化剂中,合金化或掺杂引入的电子效应、应变效应与集合效应通常相互耦合,使得分离并阐明电子结构对活性的本征贡献成为长期存在的挑战。现有研究对富电子还是缺电子Pt表面更有利于ORR尚无定论,阻碍了掺杂剂的理性设计。因此,亟需构建一个能够严格解耦电子效应与其他结构效应的模型催化剂平台,从而精准建立电子结构、中间体吸附行为与本征ORR活性之间的直接关联。
本研究通过构建定义明确的铂基纳米线(NW)模型平台,首次实现了电子效应与应变效应、集合效应的严格解耦,并建立了电子结构-吸附行为-ORR活性之间的直接关联。其核心亮点在于:1) 模型设计创新:选择给电子Re和吸电子Au分别掺杂Pt NW,在保持相同形貌、{111}表面晶面和配位环境的前提下,仅改变表面电荷密度,从而将电子贡献分离出来。2) 一致的活性趋势:从旋转圆盘电极到膜电极组件(MEA)级别,均观察到PtRe > Pt > PtAu的活性趋势,证实富电子Pt表面更有利于ORR。PtRe NW在MEA中0.9 V下质量活性达0.68 A mgPt⁻¹,超过美国能源部目标。3) 卓越稳定性:PtRe NW在30000次循环后质量活性衰减仅11.8%,电压损失12 mV,兼具高活性与强耐久性。4) 机理阐明:原位光谱与DFT计算共同揭示,Re向Pt提供电子生成富电子表面,降低氧中间体吸附能,而Au则产生相反效果。
本研究旨在阐明电子结构在调控铂基催化剂ORR活性中的本征作用,排除应变效应与集合效应的干扰。研究团队设计并合成了三种具有相同形貌(一维纳米线)、暴露晶面({111})和配位环境的模型催化剂:PtRe NWs、Pt NWs和PtAu NWs。其中,Re(电负性1.9)作为给电子掺杂剂,Au(电负性2.4)作为吸电子掺杂剂,而Pt的电负性为2.28。通过控制掺杂量(约5 at.%)并保持纳米线直径(约3 nm)和长度一致,成功构建了仅表面电荷密度不同的理想模型体系。
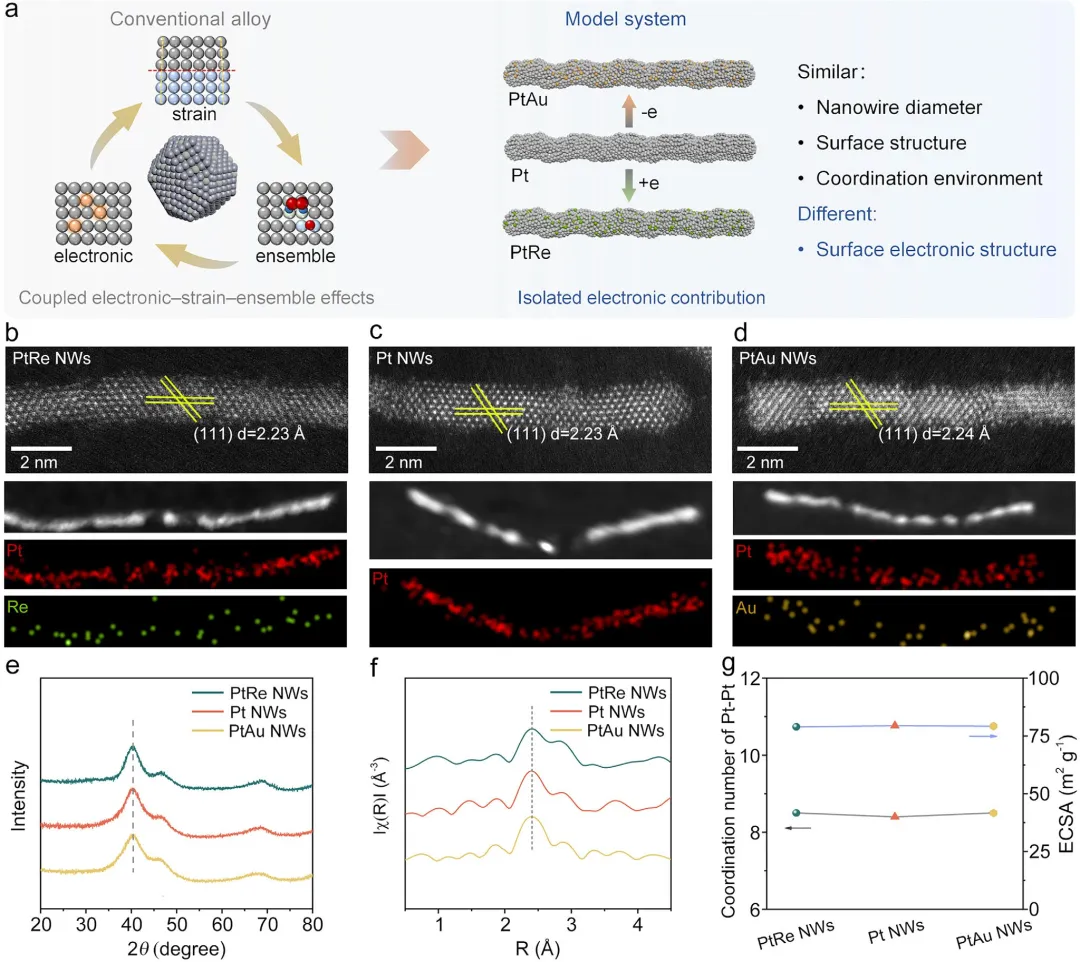
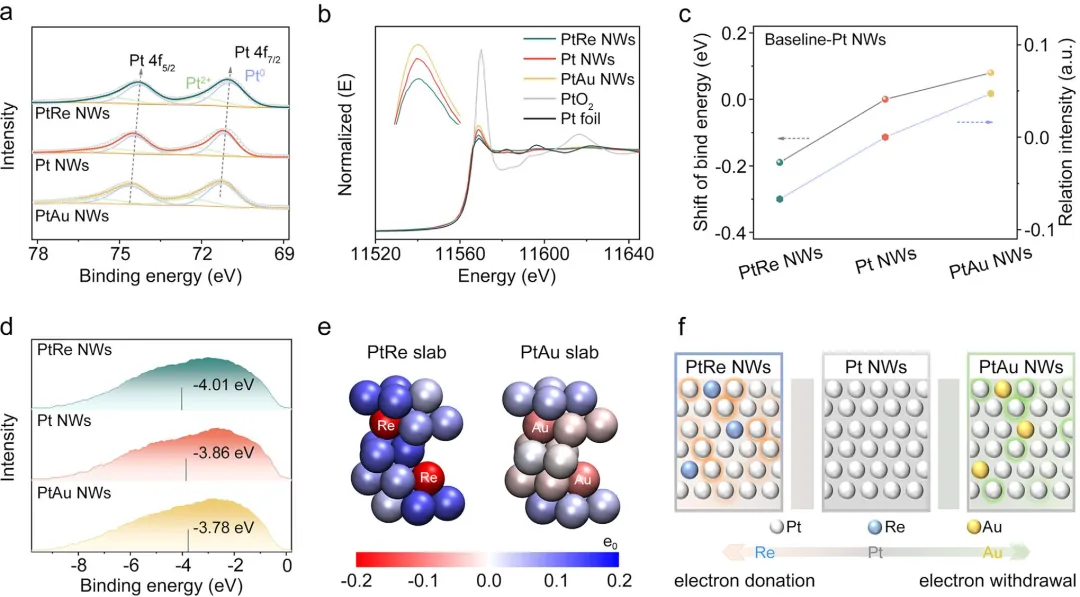
首先,通过系统的结构表征确认了模型的成功构建。 X射线衍射、透射电镜和X射线吸收谱表明,三种纳米线均具有面心立方结构,且掺杂元素均匀分布,未引起明显的晶格应变或配位环境变化。X射线光电子能谱和X射线吸收近边结构分析证实,PtRe NW中Pt的4f结合能较纯Pt NW负移(富电子),而PtAu NW中Pt的结合能正移(缺电子),验证了预期的电子转移方向。
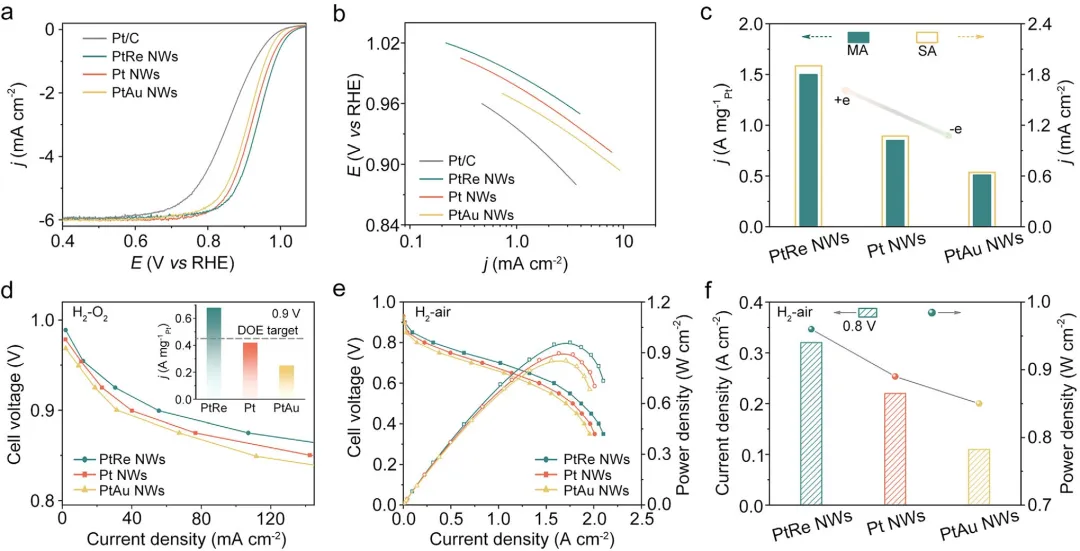
其次,在电化学性能评估方面, 旋转圆盘电极测试显示,PtRe NW的比活性为1.90 mA cm⁻²,显著高于Pt NW(1.07 mA cm⁻²)和PtAu NW(0.64 mA cm⁻²)。在MEA测试中,PtRe阴极在0.9 V下质量活性达0.68 A mgPt⁻¹,优于DOE 2025目标(≥0.44 A mgPt⁻¹)。耐久性测试表明,PtRe NW在30000圈方波循环后质量活性衰减仅11.8%,0.8 A cm⁻²下电压损失12 mV,远优于Pt NW和PtAu NW。
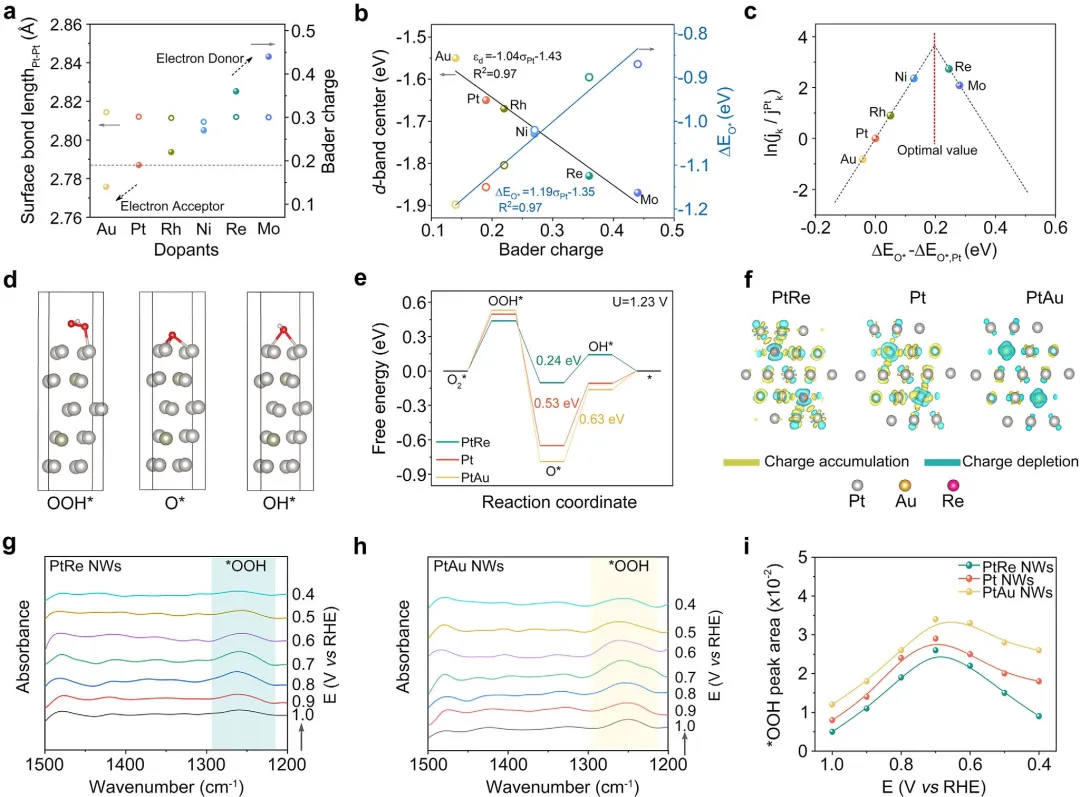
最后,通过原位表面增强红外吸收光谱和DFT计算揭示了电子效应的机理。 原位SEIRAS显示,PtRe NW表面吸附的CO峰位较Pt NW红移,表明富电子Pt表面削弱了含氧中间体的吸附强度。DFT计算进一步表明,Re掺杂使Pt的d带中心下移,降低了OH和O的吸附能,从而加速了ORR决速步的动力学。相反,Au掺杂使d带中心上移,增强了中间体吸附,抑制了反应速率。该工作通过“模型催化剂解耦电子效应 → 建立电子结构与活性定量关联 → 原位光谱与理论计算验证机理*”的研究路径,为理性设计高性能Pt基ORR催化剂提供了清晰的电子结构调控原则。
图1 PtRe NWs、Pt NWs和PtAu NWs的结构与组成表征。(a)富电子和缺电子的Pt基模型催化剂的示意图。在传统合金催化剂中,电子效应、应变效应和集合效应相互耦合。(b)PtRe NWs、(c)Pt NWs和(d)PtAu NWs的HAADF-STEM图像和STEM-EDS元素面分布图。(e)XRD谱图,(f)R空间中的傅里叶变换EXAFS谱图。(g)Pt-Pt配位数和ECSA。
图2 PtRe NWs、Pt NWs和PtAu NWs的电子结构表征。(a)Pt4f的高分辨XPS谱图。(b)PtL₃边XANES谱图。(c)结合能位移和相对强度。PtNW被选为基准。(d)高分辨XPS测得的价带谱。(e)Bader电荷分析。(f)Re/Au与Pt之间的电子转移示意图。
图3 商业Pt/C、PtRe NWs/C、Pt NWs/C和PtAu NWs/C催化剂的ORR活性与MEA性能。(a)在O₂饱和的0.1M HClO₄中的ORR极化曲线(转速1600rpm,扫描速率10mV s⁻¹。(b)Tafel斜率曲线。(c)在0.90VRHE下的MA和SA。(d)H₂-O₂燃料电池极化曲线。阴极Pt载量:0.1mgPt cm⁻²,背压:150kPaabs,80°C,以及100%RH。插图为在0.90V下的MEA MA。(e)H₂-空气燃料电池极化曲线。(f)在H₂-空气燃料电池中0.80V下的电流密度和功率密度。
图4 揭示ORR活性的调控机制。(a)不同层中Pt-Pt的表面键长和Pt的Bader电荷。(b)DFT计算确定的d带中心和ΔEO*与Pt的Bader电荷的相关性。R²,决定系数。(c)基于DFT计算,以氧吸附能为函数的模型催化剂活性火山图。(d)PtRe(111)层上ORR的吸附氧中间体构型。(e)在平衡电位(U=1.23V)下,PtRe(111)、Pt(111)和PtAu(111)层的计算自由能图。(f)PtRe(111)、Pt(111)和PtAu(111)slabs的差分电荷密度分布。(g)PtRe NWs和(h)PtAu NW在O₂饱和的0.1M HClO₄中,于1.0至0.4VRHE电位范围内ORR过程中的原位SEIRAS测量。(i)PtRe NWs、Pt NWs和PtAu NWs在1.0至0.4VRHE范围内*OOH峰面积变化。
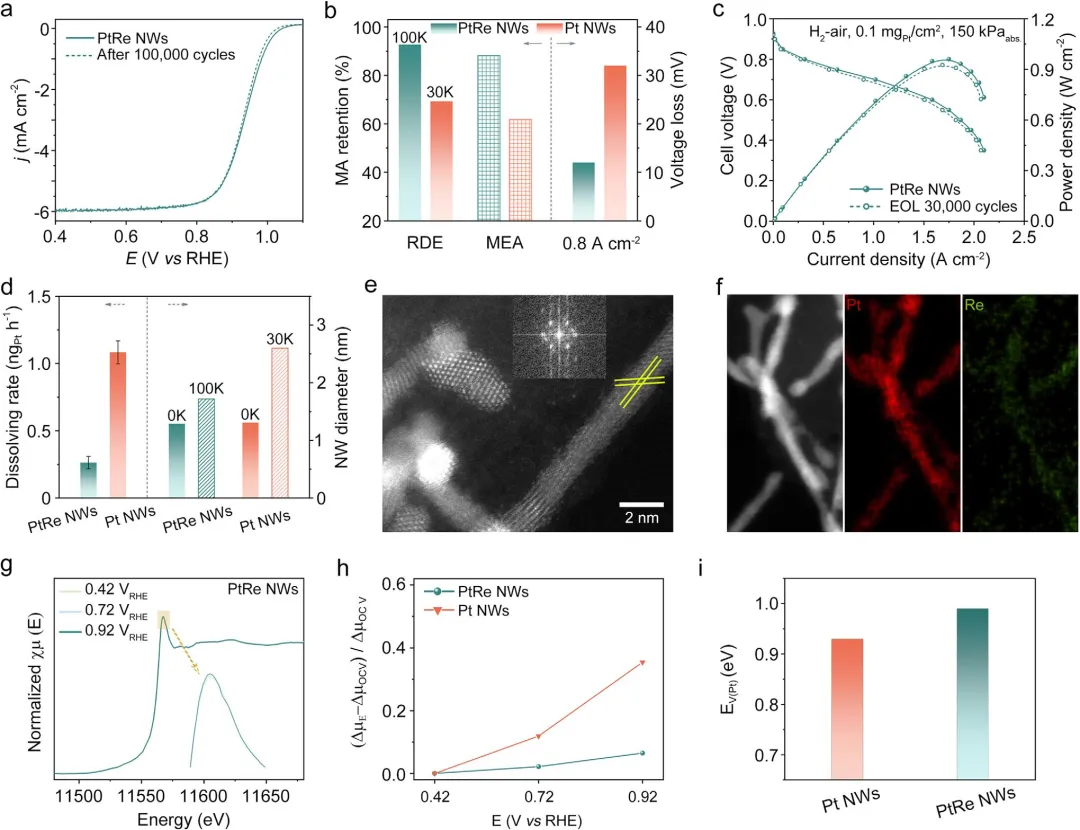
图5 PtRe NWs/C催化剂的ORR稳定性。(a)在AST 100K圈前后,O₂饱和的0.1M HClO₄中的ORR极化曲线。(b)RDE和MEA中的MA保持率以及MEA中在0.80A cm⁻²下的电压损失。(c)AST 30K圈前后的H₂-空气燃料电池极化曲线。(d)30K圈AST后Pt的溶解速率与纳米线直径的比较。(e)PtRe NW在AST 30K圈后的原子分辨率HAADF-STEM图像和(f)EDS元素面分布图像。(g)PtRe NWs在不同电位下的原位PtL₃边XANES谱图。插图为棕色矩形标记区域的放大图。(h)PtRe NWs和Pt NWs的XANES谱图中Pt吸附边峰变化(Δμ)随电位的比较。(i)Pt NWs和PtRe NWs的空位形成能。
通过整合实验与理论研究,该研究表明通过杂原子掺杂调控Pt的电子态直接决定了其ORR活性:富电子Pt增强活性,而缺电子Pt削弱活性。合成了三种直径相近的NW模型催化剂——PtRe、Pt和PtAu NWs——以分离电子效应。在RDE条件下,PtRe NWs的比活性(1.90 mA cm⁻²)高于Pt NWs(1.07 mA cm⁻²)和PtAu NWs(0.64 mA cm⁻²)。同时,在MEA条件下,在0.9 V下还实现了0.68 A mgPt⁻¹的高质量活性,超过了Pt NWs(0.42 A mgPt⁻¹)、PtAu NWs(0.25 A mgPt⁻¹)以及美国能源部(U.S. DOE)目标(≥0.44 A cm⁻²)。值得注意的是,PtRe NWs催化剂表现出强耐久性,在30K循环方波测试后,其在0.9 V下的质量活性衰减为11.8%,在0.8 A cm⁻²下的电压损失为12 mV,超过了美国能源部2025年目标(≤40%和≤30 mV)。DFT计算、XPS和XANES分析揭示,Re掺杂使Pt表面富电子,而Au掺杂则使其缺电子。这种电子调控改变了含氧中间体的吸附强度,与原位SEIRAS结果以及从RDE和MEA测量中观察到的ORR性能(PtRe NWs > Pt NWs > PtAu NWs)相一致。该研究不仅验证了富电子Pt在增强ORR活性中的关键作用,而且还为设计其他先进电催化剂提供了一种广泛适用的策略。

 点击蓝字,关注我们,每天有干货
点击蓝字,关注我们,每天有干货