『锂电』南京大学周豪慎团队Nature:电场响应靶向抗溶剂构筑梯度溶剂电解液,实现安培时级高稳定锂金属软包电池
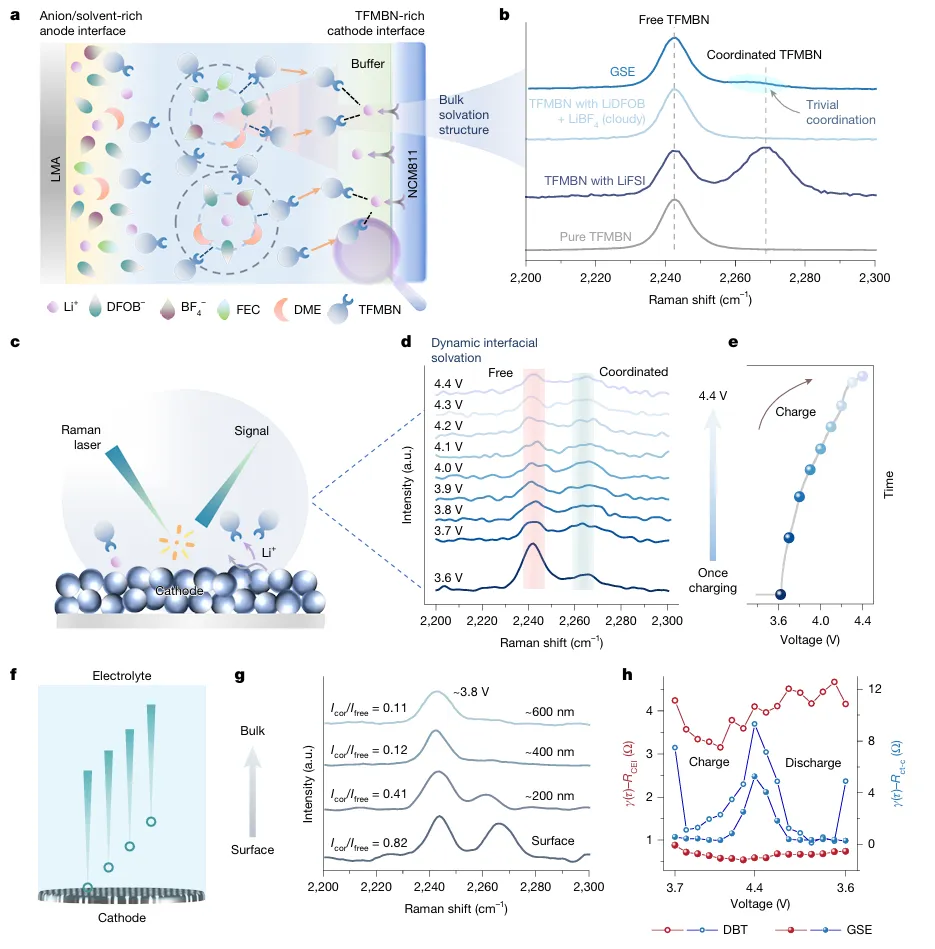
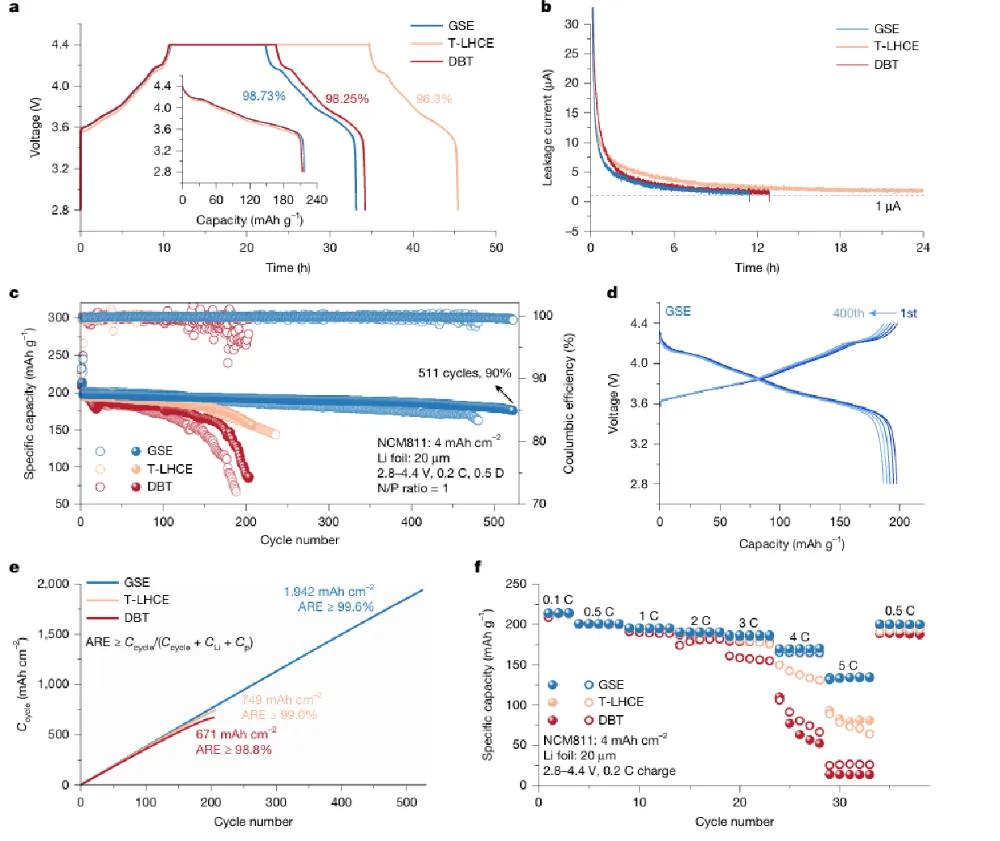
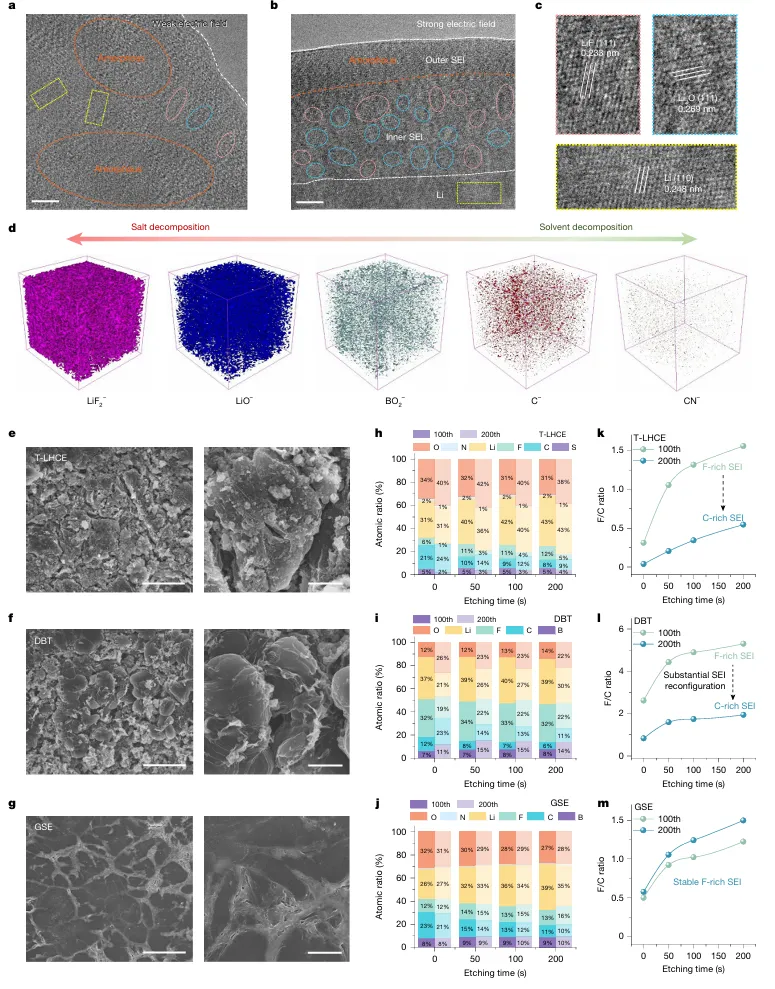
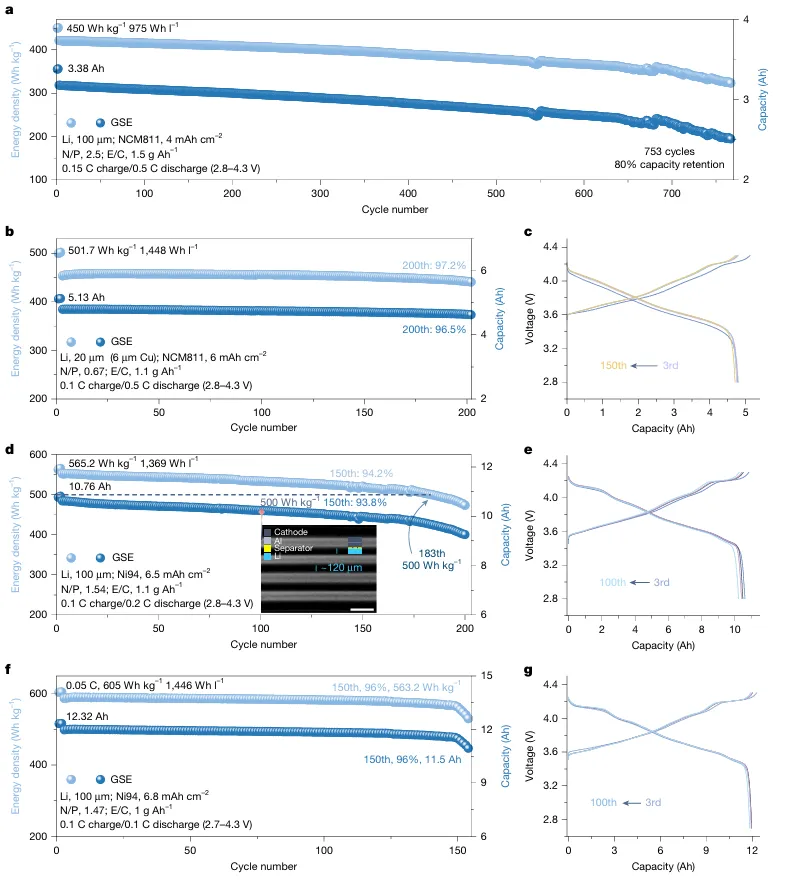
醚基局部高浓电解液(LHCE)凭借优异负极界面稳定性,是锂金属电池主流电解液体系,但高压全电池工况下存在难以根除的致命缺陷:充电时正极脱嵌大量锂离子,迫使电解液溶剂、阴离子发生脱溶剂重构,极易暴露易氧化分子,引发持续电解液分解副反应;长周期循环过程中溶剂、锂盐组分不断消耗,溶剂化结构持续劣化,正极 / 负极双界面膜 CEI、SEI 同步衰败,容量断崖式衰减。现有高浓、弱溶剂电解液仅优化静态溶剂鞘结构,完全忽略充放电强电场动态调控效应,缺少电场响应型功能分子设计思路:静态下不参与锂配位,高压充电时定向迁移至正极表面、主动捕获脱嵌锂,规避传统电解液脱溶剂氧化路径。传统改性手段无法同步解决三大痛点:1)高压下电解液大规模氧化分解;2)循环过程溶剂化不可逆重构;3)大容量薄电解液软包电芯寿命短,N/P 低配比体系失效速度更快,电解液配方开发依赖海量试错,缺少动态梯度溶剂化完整底层理论。本工作设计电场响应靶向配体抗溶剂(TFMBN),构建单相梯度溶剂电解液 GSE。静态状态下 TFMBN 配位能力弱,不会破坏阴离子富集基础溶剂鞘;4.3–4.6 V 高压强电场作用时分子定向迁移至 NCM 正极表面,氰基位点主动捕获脱嵌 Li⁺,形成正极局部缓冲溶剂层,从根源规避传统电解液溶剂大量脱溶氧化。体系采用 DME 主溶剂、LiDFOB/LiBF₄双锂盐、FEC 添加剂搭配 TFMBN 功能助剂。依托原位拉曼、空间分辨光谱、AIMD 从头动力学、冷冻 TEM、TOF-SIMS、XPS 深度刻蚀、DRT 弛豫解析、高低温扣电、450 Wh/kg、500 Wh/kg、605 Wh/kg 多规格安时级锂金属软包完成全维度验证。450 Wh/kg Li||NCM 软包 753 圈容量保有 80%;605 Wh/kg 超高能量电芯 150 圈容量留存 96%;4.6 V 超高压、80 ℃高温、长日历静置严苛工况均可稳定运行,为大容量高能量锂金属电池提供电场动态溶剂化电解液全新调控范式。①机理创新:首次提出电场诱导梯度溶剂化完整理论,区分静态 / 充电双状态分子配位行为,阐明 TFMBN 靶向缓冲层抑制电解液氧化、稳定双界面双重机制;②电解液创新:单相无分相 GSE 体系,无需高盐浓度,仅少量 TFMBN 即可实现正极局部梯度溶剂层,兼顾锂金属负极稳定阴离子鞘与高压正极抗氧化能力;③表征创新:联用空间分辨原位拉曼追踪电极到电解液本体溶剂结构演变、电场下分子动力学模拟、冷冻电镜原子尺度 SE/CEI 观测、多谱学联用解析循环界面动态重构;④工程创新:适配 1.0–1.5 g Ah⁻¹ 极低薄电解液软包工艺,开发 450~605 Wh/kg 多能量密度叠片电芯,高电压、宽温、长静置场景均具备车载产业化价值。图 1 梯度溶剂电解液 GSE 结构与电场响应分子行为a GSE 单相电解液梯度结构示意图,负极侧阴离子 / 醚富集稳定 SEI,正极 TFMBN 定向富集形成缓冲溶剂层;b 静态条件不同溶剂拉曼光谱,TFMBN 静态几乎不与 Li⁺配位;c 充电原位拉曼测试装置与 4.4 V 下 TFMBN 配位信号变化;d 充电过程激光由正极向电解液本体逐层扫描的空间分辨拉曼;e 不同深度 I 配位 / I 自由信号比值梯度变化;f 充放电 DRT 弛豫分解,正极界面阻抗长期稳定无激增1)传统 LHCE、无功能氟醚电解液充电时正极大量 DME 脱溶,直接暴露易氧化醚分子;GSE 依靠电场驱动 TFMBN 富集正极,缓冲脱嵌锂,抑制溶剂解离;2)静态电解液拉曼无配位 TFMBN 特征峰,充电升至 4.0 V 以上氰基配位峰显著增强,证明电场激活分子配位能力;3)从正极表面向电解液本体,配位 TFM 信号持续衰减,直观验证电极 - 本体梯度溶剂化空间分布;4)DRT 数据显示 GSE 正极电荷转移、CEI 弛豫峰幅值稳定,对比普通电解液循环界面阻抗持续恶化。a 4.4 V 恒压静置漏电流测试;b 不同电解液 Li||NCM 扣电 0.2 C 长循环曲线;c 千圈充放电电压平台;d 全电池平均可逆效率 ARE 统计;e 0.1~5 C 梯度倍率容量;f 2 C 高倍率长循环稳定性1)T-LHCE、普通氟醚 DBT 电解液恒压漏电流远高于 GSE,高压持续氧化副反应剧烈;2)标准 4.4 V Li||NCM(4 mAh cm⁻²、20 μm薄锂)扣电,GSE 循环 511 圈仍保有 90% 容量,对比对照组 200 圈以内大幅衰减;3)GSE 体系全电池平均可逆效率超 99.6%,DB 仅 98.8%,有效减少活性锂不可逆损耗;4)5 C 超高倍率仍维持 134.9 mAh g⁻¹,2 C 稳定循环 700 圈,动力学适配快充需求。图 3 不同电场强度下 SE 微观结构与循环界面演化a 弱电场 Cu||Li 体系冷冻 TEM(无规整无机 SE);b 强电场 NCM 体系 GSE 形成分层有序 SE;c SE 原子层放大晶格条纹;d 循环后锂负极 SEM 形貌(GSE 致密无粉化,对照组大量碎屑沟壑);e-g T-LHCE/DBT/GSE 三组 XPS 深度刻蚀 F/C 比值变化;h-j 百圈、二百圈 SE 无机 / 有机组分演变1)弱电场无充足电场驱动 TFMBN 定向富集,SE 以无定形有机组分为主,易产生死锂;4.4 V 强电场下 GSE 生成外层有机、内层LiF有序梯度 SE;2)200 圈后 T-LHCE、DBT 负极 SE 氟含量大幅下降,有机碳酸盐持续累积;GSE 全程维持高LiF占比,界面机械稳定性更强;3)TOF-SIMS 证实 GSE 循环后锂表面无机碎片信号占比高,过渡金属溶解、沉积现象被显著抑制。a 450 Wh/kg Li||NCM 6 Ah 软包循环曲线; b,c 501.7 Wh/kg 高载 N/P=0.67 超薄锂软包;d,e 565 Wh/kg Ni94 高镍电芯循环;f,g 605 Wh/kg 极限能量软包长循环1)450 Wh/kg 叠片软包电解液用量仅 1.5 g Ah⁻¹,753 圈容量留存 80%;2)N/P 低至 0.67、20 μm超薄锂 5 Ah 电芯,200 圈能量保持 96.5%;3)最高 605 Wh/kg Ni94 软包 0.05 C 低倍率首圈能量密度达标,150 圈仍维持 96% 容量保有,电芯膨胀可控。传统醚基 LHCE 电解液在高压全电池充电时会发生大规模溶剂脱溶氧化,长循环下溶剂化结构不可逆重构,正负极界面持续衰败,大容量薄电解液软包电池寿命极差。本工作开发电场响应 TFMBN 靶向抗溶剂,构建单相梯度溶剂电解液 G:静态不破坏阴离子富集锂溶剂鞘,充电强电场驱动分子定向迁移至正极表面形成缓冲层,阻隔溶剂大规模脱溶氧化,同时在锂金属侧生成富LiF致密稳定 SE 膜。GSE 体系适配 4.4–4.6 V 高压、-20~80 ℃宽温域、长日历静置严苛工况;实验室扣电 511 圈容量 90%,450~605 Wh/kg 多款工业安时级软包电芯实现超长循环,超薄低 N/P 无过量锂体系稳定性大幅领先传统局部高浓电解液。该电场动态梯度溶剂化设计思路,为高能量、大容量车载锂金属电池电解液工程化提供普适分子调控理论。Single-phase gradient-solvation-electrolyte stabilized Li metal batteries, Nature, 2026; https://doi.org/10.1038/s41586-026-10732-z本文内容来源于学术研究论文,版权归原作者所有。转载旨在分享学术成果,仅供参考,不构成任何应用建议。如涉及作品内容、版权或其他问题,请及时联系处理。


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
