Alport综合征是一种常见的遗传性肾病,问题出在COL4A3、COL4A4或COL4A5这三个基因上。它们一旦突变,肾脏基底膜的胶原结构就会受损,不少患者最终走向肾衰竭。常规的全外显子测序这类检测,通常只能找出55%–86%的病因,大量藏在非编码区和复杂结构里的变异容易被漏掉,成了诊断上的“盲区”。
近日,《自然·通讯》刊登了一篇题为“Sequential sequencing reveals the architecture and complexity of genomic variants in patients with Alport syndrome”的论文。通讯作者是南京大学医学院附属金陵医院的刘志红院士和浙江大学龚亮研究员。
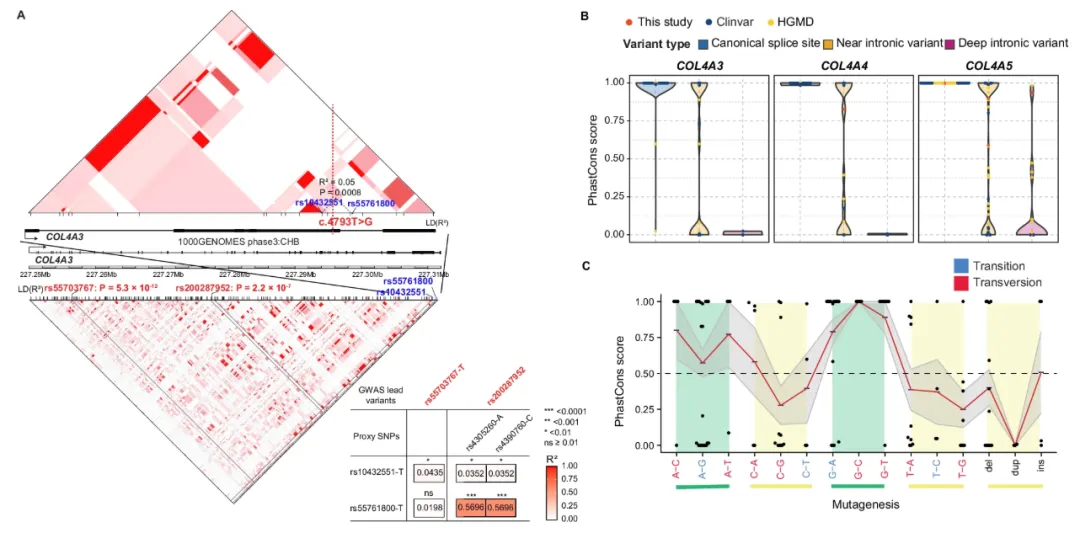
研究团队对555名经肾活检确诊的中国Alport综合征患者设计了一套层层递进的测序方案。所有人先做全外显子测序,没找到病因的再补做全基因组测序、RNA测序,最后从52名仍未确诊的患者中随机选了13人,用纳米孔长读长测序技术再过一遍。这样一步步走下来,509名患者(检出率91.7%)身上一共找到了431个不同的致病性变异。其中182个(占42.2%)是此前从未报道过的新变异,在HGMD Pro和ClinVar这些公共数据库里都查不到。这些新变异散落在三个致病基因的各个结构域上,没有明显的热点区域。
有意思的是,非编码区的变异贡献了16.2%的病因,而且有些深部内含子突变呈现出中国人特有的突变模式。传统的短读长测序容易漏掉的拷贝数变异和复杂结构变异,在本研究中占到了5.7%–9.1%。最关键的发现来自纳米孔长读长测序——团队首次在Alport综合征患者中找到了两种全新的结构变异类型。一种是大片段DNA插进内含子区域,比如一个约2千碱基的转座元件插入,它能充当弱双向启动子,扰乱基因的正常转录。另一种是涉及重复和倒位的复杂重排,直接破坏了多个外显子。正是这些“隐藏”得很深的变异,解释了很多患者临床表现典型却长期得不到基因诊断的困境。
研究还系统分析了基因型与临床表现之间的关联。比如,X连锁遗传的男性患者如果携带无义变异或结构变异,进展到肾衰竭的风险要比错义变异者高得多。听力损失和COL4A5胶原结构域的半合子变异关系密切。肾囊肿的出现则跟COL4A3杂合变异以及COL4A5半合子变异显著相关。基于这些发现,研究团队建议,Alport综合征的遗传诊断应该把全基因组测序作为一线工具;对那些常规方法查不出来的病例,纳米孔长读长测序是一个非常有价值的补充。这套多层级测序的思路,也为其他遗传性疾病的精准诊断提供了一个可推广的范例。





 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
