南京工业大学:二苯基氧膦介导的多氟(杂)芳烃选择性脱氟氢化
- 2026-05-28 03:22:48

论文信息

第一作者:黄雪莹
通讯作者:郭凯教授、李朝军教授、褚雪强副教授
单位:南京工业大学背景
多氟芳烃在医药、农用化学品、材料科学及有机合成等领域的应用(图1,A),持续受到科研人员的广泛关注,并有力推动了高效合成策略的发展。然而,传统的氟化方法在构建多氟芳烃时面临显著局限:往往依赖于预先通过多步反应安装特定官能团,或借助精心设计的导向基团来控制区域选择性。针对这一问题,利用来源广泛的全氟芳烃进行氢解反应,不仅为部分氟代芳烃的合成开辟了极具前景的新途径,同时也为深入理解碳-氟键活化的本质提供了有力工具。目前,该反应已在过渡金属催化、光催化及主族体系中得到验证,其中Weaver等报道的NaBH₄亲核芳香取代氢解是经典体系。
在磷化学领域,基于P(III)/P(V)氧化还原循环的催化氢化已有报道。然而,现有体系仍存在明显局限:Radosevich等发展的方法依赖于复杂配体以约束磷中心几何构型(图1,B,i、ii、v),而简单磷体系则需过量强还原剂实现催化剂再生(图1,B,iii-iv)。另一方面,Stephan等发现路易斯酸性P(V)盐及具有较大位阻的路易斯酸碱对虽能活化C−F键(图1,D),但反应主要局限于氟代烷烃,且鏻盐稳定性较差。受Garcia等关于简单P(III)计量氢解反应的启发(图1,C),并结合P(V)试剂氟亲和性尚未用于C(sp²)−F官能团化。作者设想,若使用简单、稳定、廉价的H-膦氧化合物[HP(O)R'R'']作为脱氟促进剂,就可以避免使用额外的过渡金属/有机催化剂、敏感的添加剂和苛刻的反应条件。
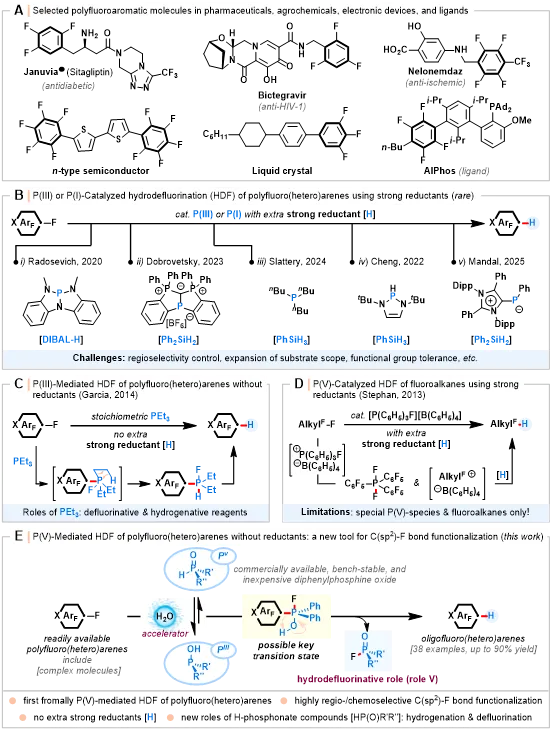
图1.多氟芳烃分子的重要性及P(I/III/V)介导的多氟(杂)芳烃脱氟氢化反应(图源:ACS Au)
Scheme 1The significance of polyfluoroaromatic molecules and P(I, III, or V)-catalyzed/mediated hydrodefluorination (HDF) of polyfluoro(hetero)arenes
概述
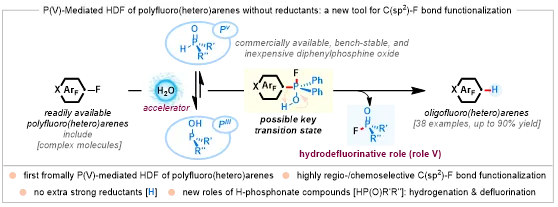
图2. 反应设计(图源:ACS Au)
Scheme 2Reaction design
近日,南京工业大学褚雪强/沈志良课题组、郭凯教授团队联合麦吉尔大学李朝军教授在JASC Au上发表题为“HP(O)Ph₂-Mediated Hydrodefluorination of Polyfluoro(hetero)arenes” 的文章。该工作的核心在于揭示了H-P(O)化合物的双重功能:其既可充当氢源,又能作为脱氟试剂参与C(sp²)–F键活化。这一策略,无需复杂P-配体结构或外部硅烷添加剂,成功克服了传统低价主族元素促进型HDF体系的内在局限,为磷酰基[P(V)]化合物在惰性化学键转化中的应用开辟了新路径。且其具优异的可放大性与官能团兼容性,不仅适用于复杂分子的后期官能化,其HDF产物还可进一步转化为其他高附加值氟化物,展现出在药物化学与有机合成中的广阔应用前景。机理研究与DFT计算表明:(1)H-P(O) →P-OH互变异构是反应的关键决速步骤,水可作为该过程的潜在加速剂;(2)反应遵循连续的氧化加成与质子转移机制。此外,进一步探索发现,H-P(O)Ph₂在C(sp²)–S及C(sp²)–P键断裂方面同样具有应用潜力。
本文要点
要点一:条件优化
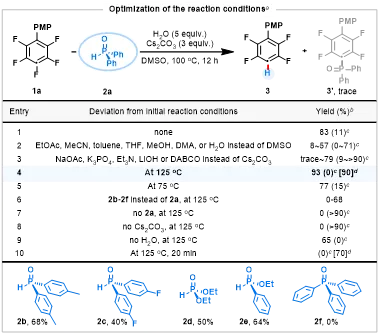
图3.反应条件优化(图源:ACS Au)
Scheme3Optimization of the reaction conditions
作者以2,3,4,5,6-五氟-4'-甲氧基-1,1'-联苯(1a)为模型底物,在二苯基氧膦(2a,1.5当量)、水(5当量)和碳酸铯(3当量)存在下,于二甲亚砜中100 ℃反应12小时,进行脱氟氢化反应研究。令人满意的是,目标产物3的核磁收率达到83%,原料1a剩余11%(Entry 1)。值得注意的是,将溶剂换为乙酸乙酯、乙腈、甲苯、四氢呋喃、甲醇、DMA或水均导致产率显著下降(Entry 2)。此外,对不同碱(如乙酸钠、磷酸钾、三乙胺、氢氧化锂和DABCO)的筛选表明(Entry 3),碳酸铯仍是最优选择。当反应温度升至125 ℃时,产物3的核磁收率提高至93%(分离收率90%),且未生成过度脱氟产物(Entry 4);而将温度降至75 ℃时,产物核磁收率为77%,原料剩余15%(Entry 5)。随后,使用不同取代基的H-磷酰化合物包括二对甲苯基氧膦2b、双(4-氟苯基)氧膦2c、亚膦酸二乙酯2d和苯基次膦酸乙酯2e)替代2a进行反应,结果显示其反应活性均有所下降(Entry 6),这归因于不同磷亲核试剂的P(=O)H→P-OH互变异构速率存在显著差异[HP(O)Ph₂>HP(O)Ph(OEt)> HP(O)(OEt)₂]。正如预期,不含H–P键的三苯基氧膦2f未能生成任何产物。对照实验进一步证实H-磷酰化合物(Entry 7)与碳酸铯(Entry 8)在反应中不可或缺。未加水(Entry 9)或缩短反应时间(Entry 10)对脱氟效果影响有限,且所有条件下直接脱氟磷酰化副产物3'的核磁收率均低于5%。
要点二:多氟(杂)芳烃化合物与氟代杂环化合物的底物范围研究
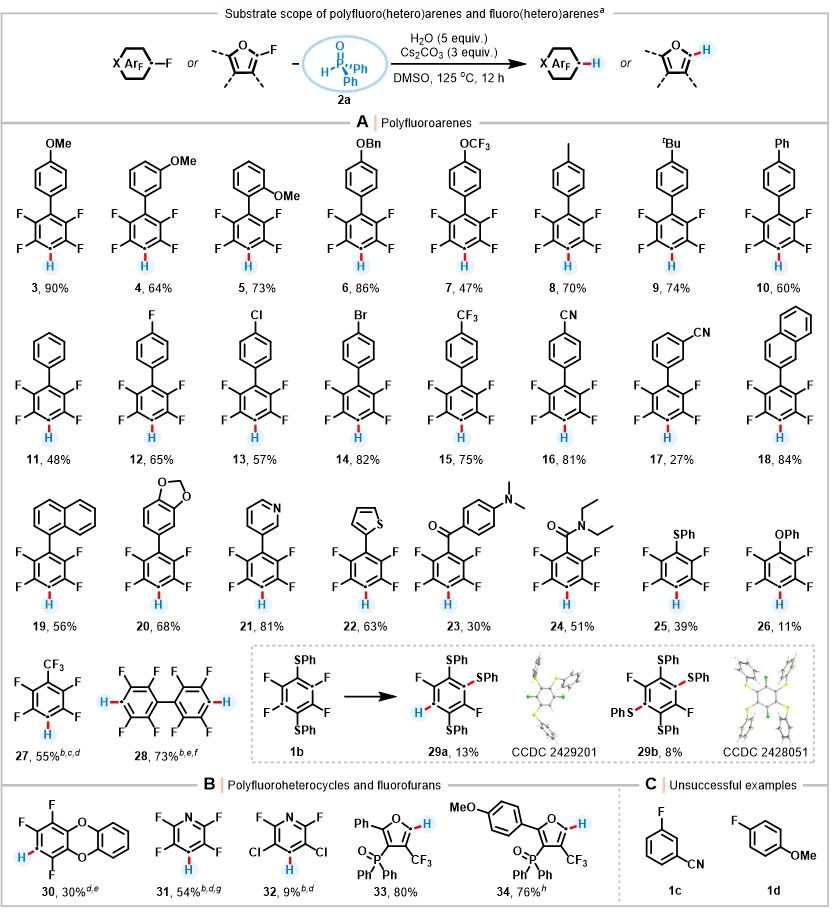
图4. 多氟(杂)芳烃化合物与氟代杂环化合物的底物范围研究(图源:ACS Au)
Scheme 4 Substrate scope of polyfluoro(hetero)arenes and fluoro(hetero)arenes
在最优反应条件下,作者将H-P(O)Ph₂介导的脱氟氢化(HDF)体系应用于多种多氟(杂)芳烃化合物及氟代杂环化合物的C(sp²)−F键选择性脱氟氢化反应。
首先,作者对一系列多氟芳烃的底物适用性进行了研究(图4,A)。带有供电子基团(产物3-9)、吸电子基团(产物12-17)[如醚基(产物3-7)、卤素(产物12-14)、三氟甲基(产物15)和氰基(产物16-17)]以及杂芳环结构[如苯并[d][1,3]二氧杂环戊烷(产物20)、吡啶(产物21)和噻吩(产物22)]的底物均可顺利实现脱氟氢化,以良好至优异收率得到相应产物。多环芳烃产物10、18和19的收率为56-84%。带有酰胺基(产物23-24)、硫醚基(产物25)和醚基(产物26)的底物,在该反应体系中均能兼容,为进一步的官能团化预留了操作空间。值得注意的是,全氟甲苯能顺利以55%的收率生成产物27,且其三氟甲基的C(sp³)–F键不会参与反应。当使用两倍量的H-P(O)Ph₂和碳酸铯时,全氟-1,1'-联苯(产物28)选择性地实现了对位两个C−F键的脱氟氢化。而使用(全氟-1,4-亚苯基)双(苯硫醚)(1b)时,则意外生成了脱氟重构芳烃产物29a和29b,其相关结构已通过X射线晶体学分析确定。
此外,在最优反应条件下(图4,B),二苯并[b,e][1,4]二噁英(产物30)和吡啶衍生物(产物31-32)也观察到了部分脱氟现象。需要指出的是,除多(杂)芳烃化合物外,单氟呋喃衍生物同样适用于该反应体系(产物33-34)。然而,对于简单的单氟芳烃如3-氟苯甲腈(1c)和1-氟-4-甲氧基苯(1d),反应未能发生(图4,C)。
要点三:药物分子的后期修饰
本脱氟氢化体系在复杂多氟芳烃类化合物的后期修饰中展现出良好的选择性与反应活性(图5),实现了雌酚酮(54%;产物35)、甲硫平(70%;产物36)、西替利嗪(62%;产物37)及紫檀芪衍生物(56%;产物38)的成功转化。

图5.药物分子的后期修饰(图源:ACS Au)
Scheme 5 Modification of complex molecules
要点四:放大反应与产物3的衍生化
随后,作者将该反应放大至3 mmol规模,以91%的分离收率获得产物3,未出现明显的效率与选择性下降,展现出优异的可放大性(图6,A)。如图6,B所示,所得产物3可作为通用合成砌块,进一步转化为一系列结构多样的氟代芳烃衍生物。在钯催化或碱性条件下,通过芳基化、烯基化、碘化、溴化及膦酸化等转化,分别以51-99%的产率得到化合物39-43。值得注意的是,上述转化过程均未使芳环中其他C(sp²)–F键断裂,保证了其结构完整性。
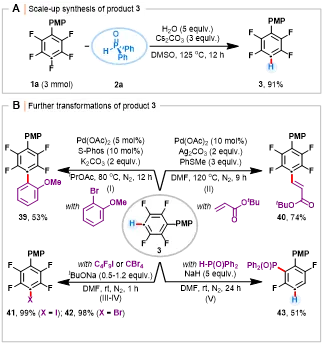
图6.放大反应和产物3的进一步转化(图源:ACS Au)
Scheme6 Scale-up synthesis and further transformations of 3
要点五:机理研究
为深入探究当前H-P(O)Ph₂介导的脱氟氢化(HDF)反应机理,作者开展了一系列对照实验,结果如图7与图8所示。首先,在自由基抑制剂[如TEMPO(2,2,6,6-四甲基哌啶-1-氮氧自由基)与1,1-二苯乙烯]存在或避光条件下进行多氟芳烃1a的脱氟官能化反应时,反应未受到显著抑制,这表明该反应可能不是自由基历程(图7,A)。
为探究H-P(O)Ph₂是否可能绕过膦诱导的HDF过程,转而通过H-P(O)化合物的质子转移途径发挥作用,作者考察了其他可作为亲核试剂或还原剂的低价膦源(图7,B)。当使用PCy₃时,得到29%的HDF产物3,同时存在未反应的1a及对位羟基化副产物3-OH(Entry 1;图7,B);而PPh₃、H₃PO₂与NaH₂PO₂则未检测到HDF产物生成(Entries 2、5、6;图7,B)。HPPh₂能够介导选择性脱氟反应,以68%的核磁收率得到产物3且无3-OH生成(Entry 3;图7,B)。ClPPh₂可使原料部分转化,生成产物3(核磁收率26%)与副产物3-OH(核磁收率7%)。且对ClPPh₂试剂的反应混合物进行¹⁹F NMR分析,检测到位于δ= -52.78 ppm处存在双重峰信号(J(FP)= 943.7 Hz)。结合文献报道可知,该信号源于P(III)中心对1a的C(sp²)–F键发生氧化加成所生成的F-P(V)化合物(Entry 4;图7,B)。这些结果表明H-P(O)Ph₂更可能是一种多功能试剂而非单纯的还原剂。
随后,作者进行了旨在捕获氟离子攫取后可能形成的多氟芳基碳负离子的对照实验(图7,C)。向模板反应中加入PhCHO、I₂、PhCH₂Br、环氧环己烷及ⁿC₄F₉I等亲电试剂后,均未得到加合物3-E(Entries 1-5;图7,C)。尽管在NaOH存在下使用ⁿC₄F₉I进行反应获得了2%的碘代产物(Entry 6;图7,C),但其可能源于HDF过程后发生的C(sp²)–H键碘化反应,如图6,B,IV所示。
接下来,作者探究了多氟芳烃取代基对反应速率的影响,并构建了哈米特曲线(图7,D)。哈米特曲线分析表明,取代基性质显著影响反应机理。当R为吸电子基团(H、Cl、CF₃、CN)时,数据呈现良好的线性相关性(ρ= -0.28);而当R过渡至供电子基团(ᵗBu、OMe)时,曲线出现明显拐点,表明反应机理发生转变。这一非线性行为更符合异步的键断裂/形成过程或完全分步的机理,其中键断裂的确切程度可能由取代基的电子性质决定。
根据图7,E所示结果,在乙腈与DCE溶剂体系中添加DMSO未对反应活性产生影响,表明DMSO与多氟(杂)芳烃发生相互作用进而导致脱氟氢化的可能性较低。
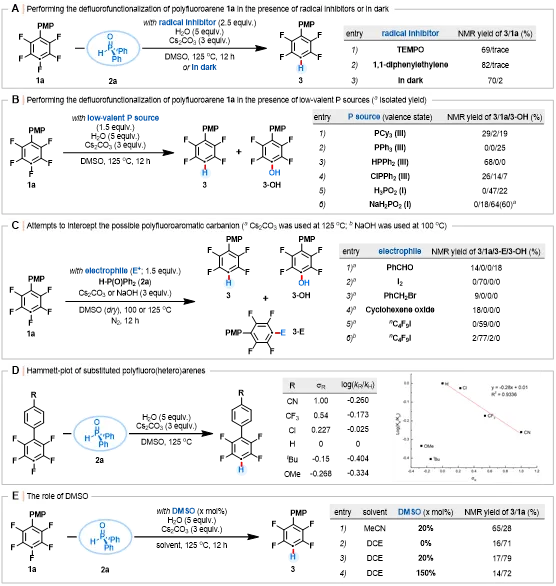
图7.机理研究(图源:ACS Au)
Scheme7 Mechanistic studies
为明确质子来源,作者开展了氘代标记实验(图8,F,式1)。使用5当量D₂O时未观察到氘代产物(图8,F,1),而10当量D₂O则能以79%收率获得脱氟氢化产物3,其氘代率为27%(图8,F,2)。在DMSOd₆中进行反应(图8,F,3)及额外添加D₂O的共反应体系(图8,F,4)分别将氘代率提升至52%与75%。进一步添加DP(O)Ph₂(2aD)可使氘代产物3D的收率达到78%,氘代率达到98%(图8,F,5)。然而,在非氘代DMSO中使用2aD/D₂O或2aD/H₂O组合的反应则呈现较低的氘代率(分别为11%与0%,图8,F,6、7)。尽管这些结果与“产物3中的质子源自H₂O和/或H-P(O)Ph₂”的推论存在矛盾,但氘富集度分析提供了一定支持:当使用D₂O和/或DP(O)Ph₂时,试剂中9.5%与6.5%的氘富集度分别导致产物出现27%与11%的氘代率(图8,F,式1,2、6)。对于在DMSOd₆中进行的反应,高达93.5%与98.6%的氘富集度却仅对应52%与75%的较低氘代率(图8,F,式1,3、4)。由此可推断,H-P(O)Ph₂与D₂O或DMSOd₆之间很可能发生了快速的H/D交换。作者通过HP(O)Ph₂的氘代标记实验验证该假设:如预期所示,在D₂O或DMSOd₆存在下处理2a,1分钟内即检测到中等程度的氘代(图8,F,式2、4);值得注意的是,该氘代过程在室温下可实现55%的氘代率(图8,F,式5)。此观测结果与前述反应体系中52%的氘代率(图8,F,式1,3)相符,从而印证了HP(O)Ph₂向DP(O)Ph₂的关键转化。此外,HP(O)Ph₂与D₂O在12小时后H/D交换率达99%,表明该步骤可能为决速步骤(图8,F,式3)。
为确认HP(O)Ph₂独特的脱氟氢化作用,作者通过核磁共振波谱监测反应进程(图8,G-1)。反应1分钟后检测到δ= 40.72 ppm处的双重峰(J(PF)=1017.7 Hz),该信号归属脱氟产物FP(O)Ph₂(2aF),其来源于HP(O)Ph₂(δ= 20.38 ppm)对多氟芳烃中氟原子的攫取,这证实了HP(O)Ph₂的核心脱氟功能。随着反应进行,HP(O)Ph₂与中间体2aF迅速消耗,在碱性条件下最终转化为二苯基次膦酸阴离子(即化合物2aOCs,δ=14.95 ppm)。
同时,作者通过¹⁹F NMR原位监测1a与2a在反应初始17分钟内的反应历程,以探究水对反应的影响(图8,G-1)。值得注意的是,添加H₂O(5当量)可同时加速反应速率并提高产物3的收率,该促进作用与HP(O)/POH互变异构的速率相关。FP(O)Ph₂在2分钟时达到最大收率(37%),此时多氟芳烃1恰好消耗完毕。当使用可将互变异构能垒从60 kcal/mol降至13.5 kcal/mol的双环胍(BG)催化剂时,反应速率进一步加快,并能高效生成3。这些发现表明:(1)H₂O可能作为HP(O) → POH互变异构的促进剂;(2)互变异构步骤可能构成关键决速步。
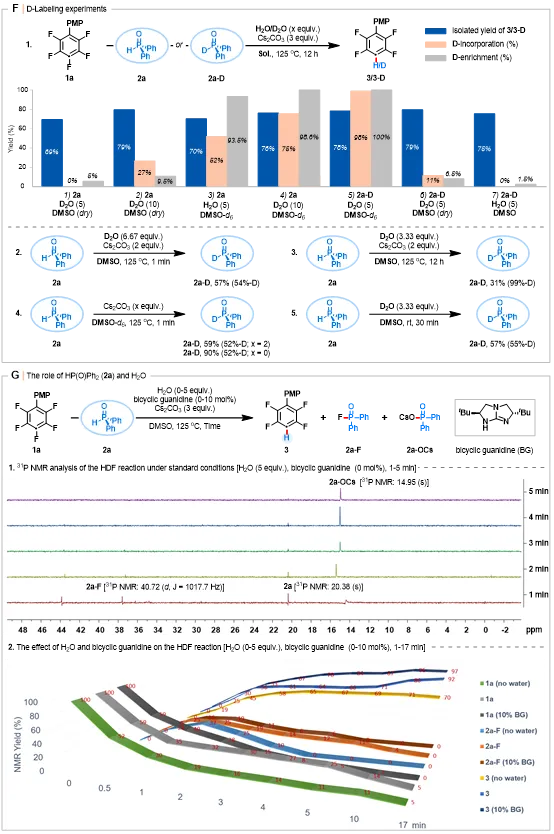
图8 机理研究(续图)(图源:ACS Au)
Scheme8 Mechanistic studies (continued)
要点六:推测的反应机理
根据上述机理研究及已有文献报道,作者提出四种可能的反应机理(图9)。
首先,二苯基氧膦2a存在三价与五价形态的互变异构平衡,该平衡通常偏向于反应活性相对较低的五价形态。值得注意的是,在该反应中水可加速二苯基氧膦2a通过质子转移互变为P(III)异构体2a',后者可能是真正的活性中间体。基于此,作者提出路径A:2a'首先通过五元环过渡态C与多氟芳环的C(sp²)–F键相互作用;随后经历质子化/脱氟过程,伴随C–F/O–H键的断裂与P–F/C–H键的形成;最终脱除F-P(O)Ph₂(2a-F),生成目标HDF产物。
与此同时,作者无法排除磷酰基阴离子(A)介导的A-T型脱氟可能性,该路径首先生成芳基阴离子E,随后阴离子被水或二甲亚砜快速质子化释放HDF产物(路径B)。此外,由1a对P(III)OH化合物2a'氧化加成生成P(V)OH中间体G(路径C),其经分子内质子转移可进一步生成F-P(O)Ph₂(2a-F)与HDF产物。
值得注意的是,尽管上述路径均涉及P(III)化合物,但实验结果表明,HP(O)R'R''与H₂O组合展现出的非常规HDF反应活性,并非源于其传统磷酸化作用。事实上,多氟芳烃与过量亲核性磷酰基阴离子的磷酸化反应可被有效抑制(路径D),这表明阴离子介导的路径并非主导。后续的哈米特研究也为这一推断提供了佐证,进一步证实了路径A在该体系中的非主导性。
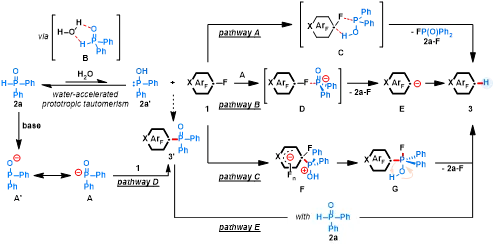
图9.可能的反应机理(图源:ACS Au)
Scheme9 Proposed mechanism
要点七:密度泛函理论(DFT)计算
为深入理解HP(O)Ph₂介导的脱氟氢化过程,作者采用密度泛函理论(DFT)对可能的反应路径进行了研究。计算结果表明,反应活性物种P(III)-OH中间体2a'首先对多氟芳烃1a中的C(sp²)–F键进行氧化加成,经由过渡态TS1(ΔG‡ = 37.5 kcal/mol)生成五价P(V)-OH中间体G。随后,中间体G通过四元环过渡态TS2(ΔG‡ = 19.8 kcal/mol)发生分子内质子转移,最终生成目标脱氟氢化产物3。该计算所揭示的分步反应机制,与哈米特研究得出的结论相吻合。
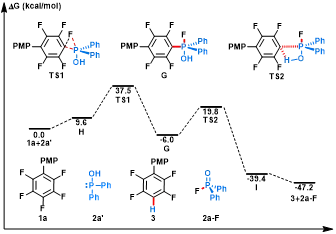
图10.H-P(O)Ph₂介导的多氟(杂)芳烃脱氟氢化反应的吉布斯自由能剖面图(图源:ACS Au)
Scheme10 Gibbs free energy profiles for H-P(O)Ph₂-mediated hydrodefluorination of polyfluoro(hetero)arenes
要点八:H-P(O)Ph₂在惰性化学键活化中的应用潜力
基于上述成功案例,作者探究了PhSiH₃存在下使用催化量2a时H-P(O)Ph₂介导多氟(杂)芳烃HDF反应的应用潜力(图11,A)。然而,目标产物3仅获得14%的核磁产率,且原料1a发生显著分解。此外,在标准反应条件下进行了H-P(O)Ph₂介导的2,2,2三氟1苯基乙酮(44)中C(sp³)–F键活化反应(图11,B)。该反应导致CF₃COPh发生降解,同时生成单脱氟产物45及一种未明确的氟化物。对于含有两个苯硫基取代基的底物1b,H-P(O)Ph₂可诱导C–S键断裂,产生的碎片在底物上发生重排,推测其可能通过游离硫醇盐的竞争性SNAr途径进行。为规避此问题,作者预先合成了单苯硫基取代物46a,并将其置于H-P(O)Ph₂介导的体系中反应,得到HDF产物3(39%核磁收率)和重组产物47a(24%核磁收率,20%分离收率)(图11,C,式1)。
值得注意的是,H-P(O)Ph₂的键活化能力并不局限于C(sp²)–S键,其同样适用于C(sp²)–P键的裂解,而此类过程在传统方法中往往需要过渡金属的参与(图11,C,式7)。但当前体系对多氟芳烃化合物46b-46f中的C–O/N/C键(图11,C,式26)、杂环48中的C(sp²)–S键(图11,D)以及酮类化合物50-51中的C(sp²)–Br/Cl键(图11,E)均未表现出反应活性。
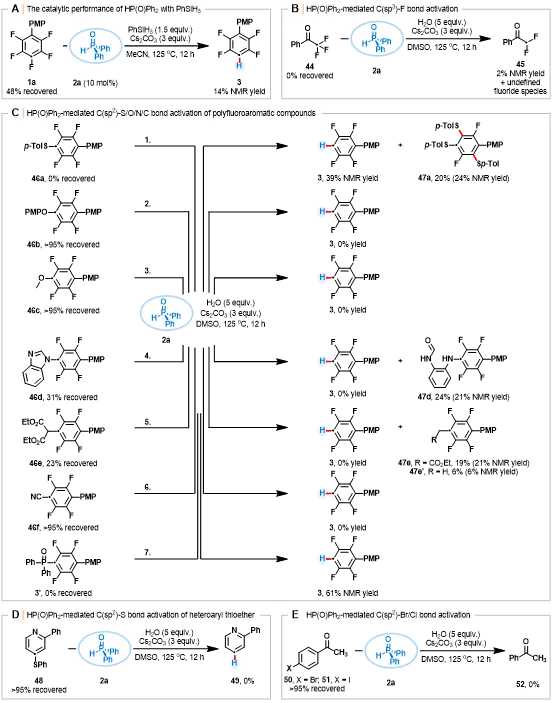
图11.HP(O)Ph₂在惰性化学键活化中应用潜能的深度评估(图源:ACS Au)
Scheme11 Further assessment of the potential of H-P(O)Ph₂for inert chemical bond activation
论文信息
Xue-Ying Huang, Xian-Xian Mao, Chenshu Dai, Ming-Yang Gu, Danhua Ge, Zhi-Liang Shen, Kai Guo, Chao-Jun Li, and Xue-Qiang Chu.HP(O)Ph₂-Mediated Hydrodefluorination of Polyfluoro(hetero)arenes. JACS Au, 2026, 10.1021/jacsau.5c01460

| 点击即可阅读合集 | ||
| 催化化学 | ||
| 分析化学 | ||
| 生物化学 |

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 清明南京亲子轻户外|住宿这样选更稳妥
- 祝贺!大橡科技&南京市中医院“类器官芯片创新中心”成立
- 南京路步行街新康大楼商办综合体出售
- 致红南京:致我们终将告别的青春与传奇
- 南京|最高可获50万元资助!2026年南京创业政策申报指南来了
- 中国“第5个直辖市”持续竞争角逐激烈:南京成都无缘,四市呼声最强!
- 现在价格168万 南京西路甲级写字楼 业主出国急售物业名称:南证大厦(又称仲益大厦)74.48平 单价2.26万/平米、
- 南京鼓楼医院集团仪征医院招聘
- 南京家教|3月25日南京家教|线上线下家教|专职在职老师家教一对一攻略|大学生家教兼职|线上线下家教老师辅导
- 南京 | 东箭道:总统府旁边的小巷,一巷看十朝
