南京大学黄小强&厦门大学「国家级高层次人才」王斌举&深研院周佳海最新Nature Catalysis | 协同双催化不对称构建C(sp³)-C(sp³) 键!
- 2026-06-13 17:23:31
南京大学黄小强&厦门大学「国家级高层次人才」王斌举&深研院周佳海最新Nature Catalysis | 协同双催化不对称构建C(sp³)-C(sp³) 键!
该系统将简单的肉桂醛转化为富含对映体的羧酸,这些羧酸带有有价值的β-或β, γ-C(sp3)手性中心,这是一种用常规方法难以实现的“非天然”转化。 通过定向进化,作者精确地控制了烷基自由基,实现了立体选择性的C(sp3)–C(sp3)键形成(38个例子,对映体过量最高达96%,非对映异构体比例最高达91:9)。 研究表明,重塑另一类硫胺素依赖性酶可以扩展自由基生物催化的范围。现代自由基化学为C(sp3)-C(sp3)键的形成开辟了一条强有力的策略,但自由基的使用在化学、非对映和对映选择性的控制方面仍然存在挑战。例如,C-自由基与羰基取代烯烃(Michael受体)的自由基共轭加成,也称为Giese反应,已被充分证明可用于合成远离羰基的C-C键。然而,通常需要双齿配位底物来控制前手性烯烃的立体化学。与化学催化剂配位的前手性Michael受体通常能得到更好的控制,但前手性C-自由基尚未得到有效调控。换句话说,在化学催化的Giese型自由基加成中,实现非对映选择性更具挑战性。 酶在选择性和可进化性方面具有独特优势,使其成为调控自由基极具吸引力的催化剂。作者质疑酶是否能提供一种可行的解决方案,以高对映和非对映选择性实现自由基C(sp3)-C(sp3)键的形成。理想情况下,应能直接利用简单的单齿烯烃,如肉桂醛。在此背景下,Melchiorre和Saravanan课题组独立地利用光照I类醛缩酶实现了肉桂醛的Giese型自由基偶联。这证明了酶控制共价结合的烯烃底物立体化学的能力。此外,Melchiorre的研究表明,(R)-2-苯基丙酸等对映纯自由基前体的手性可以通过酶活性位点中的弱相互作用得以保留。尽管取得了这些成就以及其他酶促C(sp3)-C(sp3)键形成的显著例子,但使用外消旋自由基前体实现对映和非对映选择性控制仍然是一个挑战。 在本文中,作者报道了一种对映选择性(在某些情况下也是非对映选择性)的自由基C(sp3)-C(sp3)键形成反应,该反应由一种机理上独特的双重光生物催化系统实现。该系统包含一个有机光氧化还原催化剂和一个硫胺素二磷酸(ThDP)依赖性酶。在生命系统中,ThDP酶通过酶促Breslow中间体激活醛或酮酸底物,通过双电子umpolung途径形成C(sp2)-C键。最近,作者课题组和其他人已成功改造来自荧光假单胞菌(Pf BAL)的ThDP依赖性苯甲醛裂合酶,以实现芳香醛的对映选择性ipso-C(sp2)-C(sp3)自由基偶联。在作者继续努力开发“非天然”自由基生物催化的过程中,作者试图通过将α,β-不饱和醛的β-位作为前手性C(sp3)手性中心,来实现更具挑战性的远程自由基C(sp3)-C(sp3)键形成。然而,在作者之前基于Pf BAL催化的ipso-C(sp2)-C(sp3)形成的研究中,α, β-不饱和醛不能作为酰基自由基供体被耐受。此外,Müller及其同事的一项开创性报告表明,当α, β-不饱和醛在天然的双电子缩合反应中用作酰基供体时,ThDP依赖性酶天然地只提供对β-位的1, 2-加成而非1, 4-加成。 在本文中,作者引入了一种光诱导的自由基机制,并改造了另一种不同的ThDP依赖性酶,即来自恶臭假单胞菌的苯甲酰甲酸脱羧酶(PpBFD),从而解锁了具有挑战性的C(sp3)-C(sp3)键形成反应。这种反应性不仅扩展了硫胺素依赖性酶的反应范围,而且还可以作为对化学催化N-杂环卡宾(NHC)自由基催化的补充。 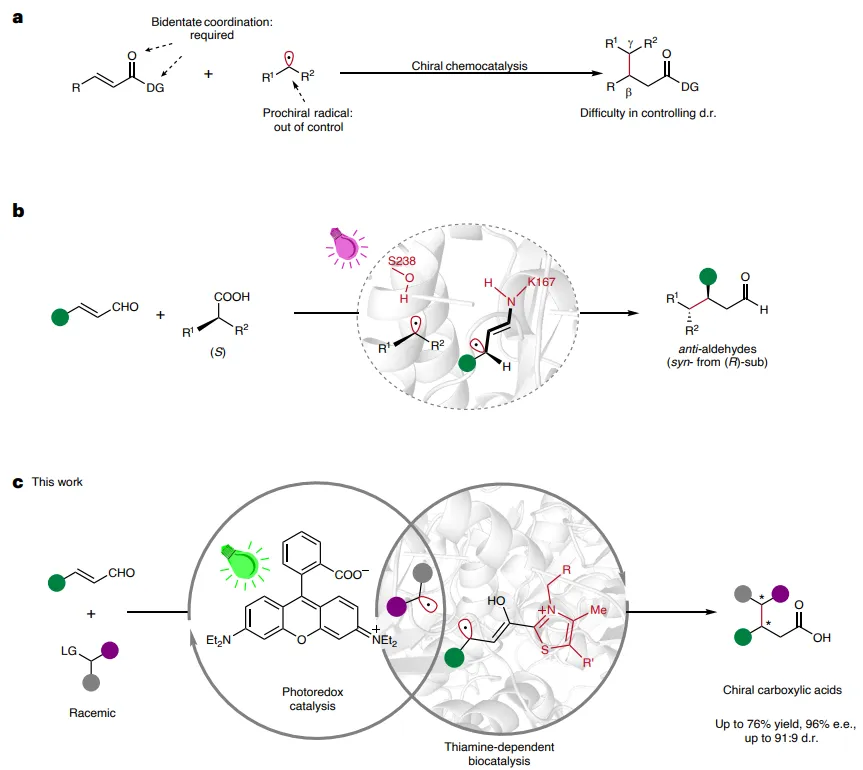
图1:利用简单肉桂醛进行C(sp3)-C(sp3)键形成的酶改造。(a)Giese型自由基C(sp3)-C(sp3)键形成的挑战。(b)直接光照I类醛缩酶以实现立体特异性自由基-自由基交叉偶联。(c)协同硫胺素依赖性酶促和光氧化还原催化以实现立体选择性C(sp3)-C(sp3)键形成。DG,导向基团;LG,离去基团。 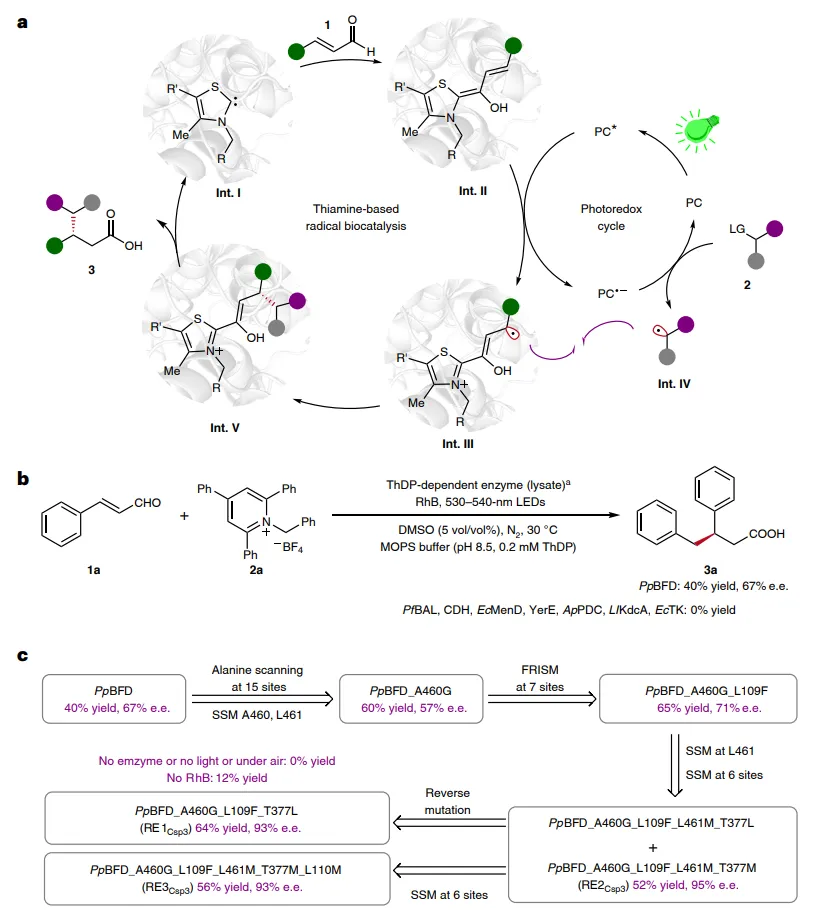
图2:设计与开发。(a)重新设计的催化循环。(b)硫胺素二磷酸(ThDP)依赖性酶的筛选。(c)PpBFD(PDB 1MCZ)的进化轨迹,以及每个变体的产率和对映体过量(e.e.)值,以及对照实验。标准条件:肉桂醛1a(0.012 mmol),Katritzky盐2a(0.004 mmol),酶(裂解液,2.5 mol%),罗丹明B(RhB,5 mol%),在50 mM MOPS缓冲液(pH 8.5,含2.5 mM MgSO4和0.2 mM ThDP)中加入5 vol/vol% DMSO,在N2气氛下于30 °C用530-540 nm LED照射搅拌20小时;反应总体积为2.5 ml。产率通过UPLC测定,并以2a为1当量基准。e.e.值通过手性固定相HPLC分析,在酯化衍生化后测定。a细胞裂解液通过超声处理获得,随后通过SDS-PAGE半定量。SSM,位点饱和诱变;FRISM,聚焦理性迭代位点特异性诱变。 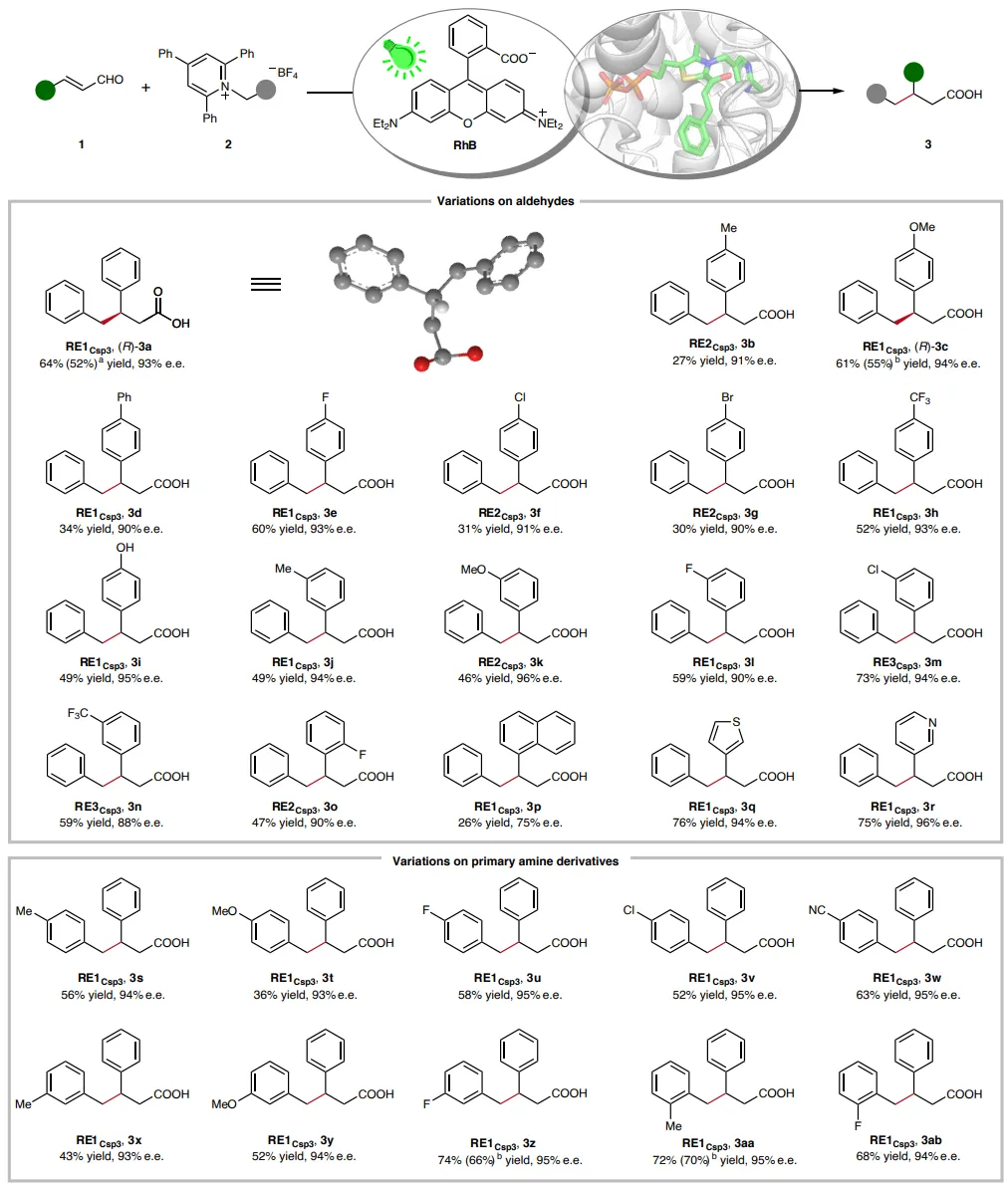
图3:具有一个手性中心的光生物催化C(sp3)-C(sp3)键形成的底物范围。除非另有说明,反应均在标准条件下(Fig. 2b)进行。产物产率通过UPLC或GC定量。e.e.值在酯化衍生化后通过手性固定相HPLC分析。绝对构型直接从3a和3c的单晶结构确定。a由0.4 mmol规模反应确定的分离产率。b由0.2 mmol规模反应确定的分离产率。 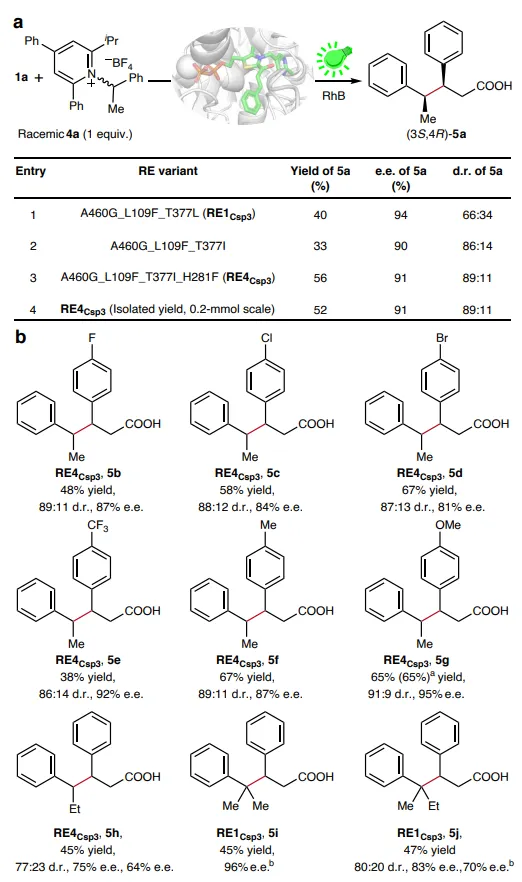
图4:使用仲和叔苄基自由基的立体选择性C(sp3)–C(sp3)键形成。(a)Katritzky盐4a的反应进化。(b)底物范围研究。除非注明,1(0.012 mmol),Katritzky盐4(0.004 mmol),RE4Csp3(2.5 mol%,裂解液),RhB(5 mol%),在50 mM MOPS缓冲液(pH 8.5,含2.5 mM MgSO4和0.2 mM ThDP)中加入5 vol/vol% DMSO,在氮气气氛下于30°C用530-540 nm(LED)照射搅拌20小时。反应总体积为2.5 ml。产率通过UPLC定量,并以4a为基准。d.r.值通过UPLC和LC-MS确定,e.e.值通过未衍生化样品的直接分析或经酯化衍生化后的手性固定相HPLC确定。a由0.2 mmol规模反应确定的分离产率。b使用N-(酰氧基)邻苯二甲酰亚胺代替Katritzky盐。 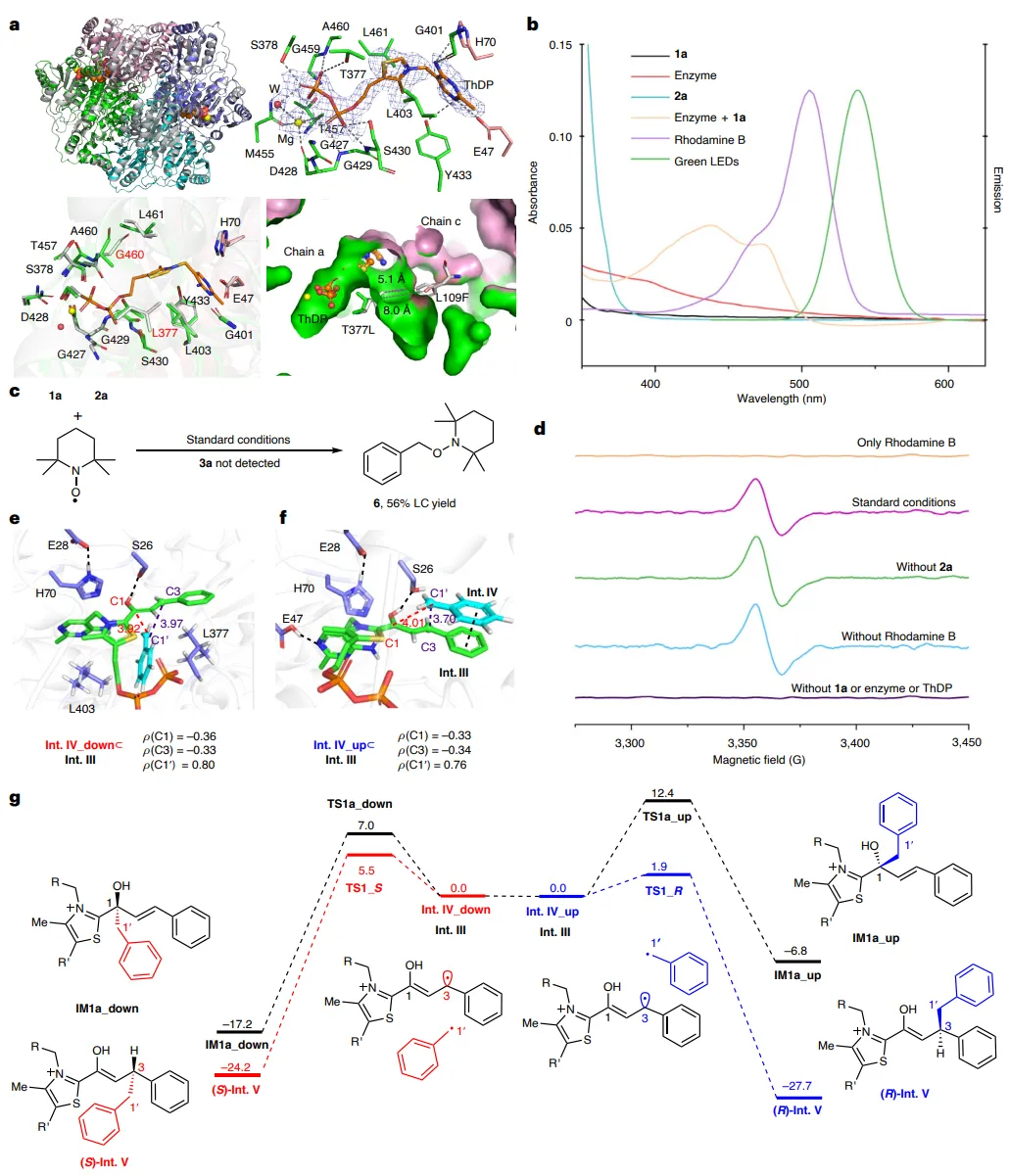
图5:机理研究。(a)野生型PpBFD(彩色)与RE1Csp3(灰色)的叠加(r.m.s.d.为0.198 Å)。(b)不同混合物的UV-vis吸收光谱和所用LED的发射光谱。(c)TEMPO捕获自由基。(d)低温EPR光谱。(e)MD模拟的Int. III⸦Int. IV向下构象。(f)MD模拟的Int. III⸦Int. IV向上构象。g,QM(B3LYP-D3/B2)/MM计算的能量分布图。Int. III⸦Int. IV代表两种前手性碳自由基的酶结合复合物。为清晰起见,省略了蛋白质支架。距离单位为Å,能量单位为kcal mol-1。 综上,作者介绍了一种协同光氧化还原催化与硫胺素依赖性酶(苯甲酰甲酸脱羧酶, PpBFD)的新型光生物催化策略,用于实现高对映选择性的C(sp3)–C(sp3)键形成。通过将简单的肉桂醛与烷基自由基前体偶联,并结合定向进化技术改造PpBFD,该系统成功构建了含有β-或β,γ-手性中心的非天然羧酸产物。 该研究成功地将自由基化学与生物催化相结合,克服了传统方法在控制两个前手性自由基偶联时的立体选择性难题,实现了优异的对映和非对映选择性(最高96% ee, 91:9 dr)。本工作不仅拓展了ThDP依赖性酶在“非天然”自由基反应中的应用范围,为复杂手性分子的绿色合成提供了新工具,而且其策略有望应用于药物化学中高价值手性砌块的高效、可持续制备,以及推动新型生物杂合催化体系的发展。 Enantioselective C(sp3)-C(sp3) bond formation by synergistic thiamine-dependent radical biocatalysis and photoredox catalysis. Nat. Catal., (2026).https://doi.org/10.1038/s41929-026-01515-w. #黄小强#王斌举#周佳海#南京大学#厦门大学#中国科学院深圳先进技术研究院#Nature子刊#催化 

自由基C(sp3)-C(sp3)键的形成已成为构建富含C(sp3)手性中心分子的一种有前景的策略。然而,实现两个前手性烷基自由基的化学和对映选择性重组仍然是一个巨大的挑战。
2026年4月2日,南京大学黄小强、厦门大学王斌举、中国科学院深圳先进技术研究院周佳海在国际知名期刊Nature Catalysis发表题为《Enantioselective C(sp3)–C(sp3) bond formation by synergistic thiamine-dependent radical biocatalysis and photoredox catalysis》的研究论文,Jianlin Chun、Yuyan Bao、Qiaoyu Zhang为论文共同第一作者,黄小强、王斌举、周佳海为论文共同通讯作者。
在本文中,作者通过将一种硫胺素依赖性的苯甲酰甲酸脱羧酶(PpBFD)与光诱导的自由基过程协同作用,解锁了非天然的光生物催化C(sp3)-C(sp3)键形成。
本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。

