【ACS Catal.】南京工业大学毛建友/宾夕法尼亚大学Patrick J. Walsh /南开大学许秀芳团队:异双金属碱催化提升1,4-加成前亲核试剂pKa极限
- 2026-05-09 13:32:07

导语
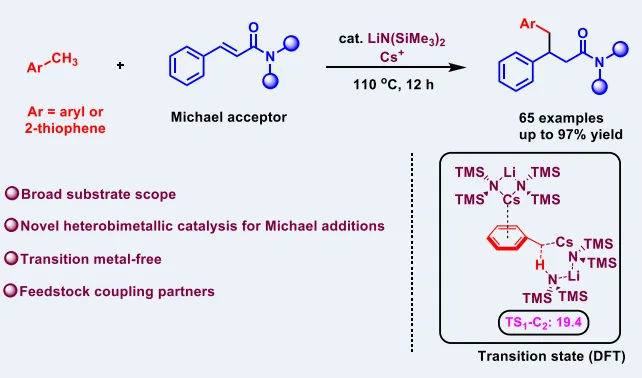
布朗斯特碱催化C−H键对碳碳双键的加成是构建C−C键的高效策略,具备优异的原子经济性。但该类催化反应需平衡碱强度、亲电双键活性与中间体碱性,高pKa底物的催化去质子化1,4-加成仍极具挑战。当前,极弱酸性C−H键(pKa>40)对迈克尔受体的催化去质子化加氢烷基化反应鲜有报道,强碱性试剂易引发副反应,且中间体难以完成催化循环。若能将适用前亲核试剂的pKa范围提升5个数量级,便可绕过传统需制备的对空气敏感有机金属试剂与过渡金属催化剂。近日,南京工业大学毛建友团队、南开大学许秀芳团队与宾夕法尼亚大学Patrick J. Walsh团队合作,开发了LiN(SiMe₃)₂/Cs⁺催化体系,成功实现甲苯、2-甲基噻吩等pKa>40的极弱酸性前亲核试剂与α,β-不饱和酰胺的1,4-加成反应。DFT计算揭示了Li⁺/Cs⁺异双金属碱的结构与协同作用机制,凸显了金属特异性阳离子-π相互作用的关键意义。相关成果发表在ACS Catalysis上(DOI: 10.1021/acscatal.6c01195)。
图文解析
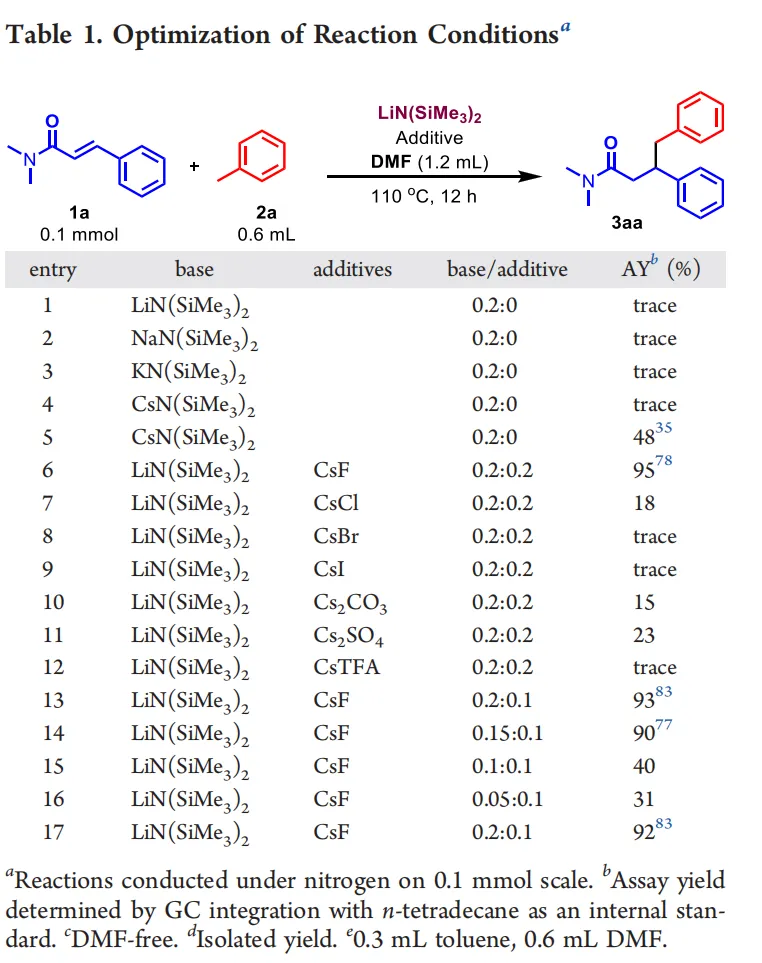
首先,作者以N,N-二甲基肉桂酰胺与甲苯为模型底物对反应条件进行系统筛选。单一碱LiN(SiMe₃)₂、NaN(SiMe₃)₂、KN(SiMe₃)₂与CsN(SiMe₃)₂都难以获得目标产物,溶剂筛选表明DMF可显著提升转化效率。作者进一步考察了LiN(SiMe₃)₂与不同铯盐的组合后发现,CsF为最优铯源,可使模版反应的GC产率提高到95%,分离产率为78%。经过对催化剂用量、底物与溶剂量等条件的细致优化,最终确定标准条件为:20 mol% LiN(SiMe₃)₂和10 mol% CsF为催化剂、DMF为溶剂,在110 ℃下反应12小时,以83%的收率得到了目标产物3aa。
(图源:ACS Catal.)
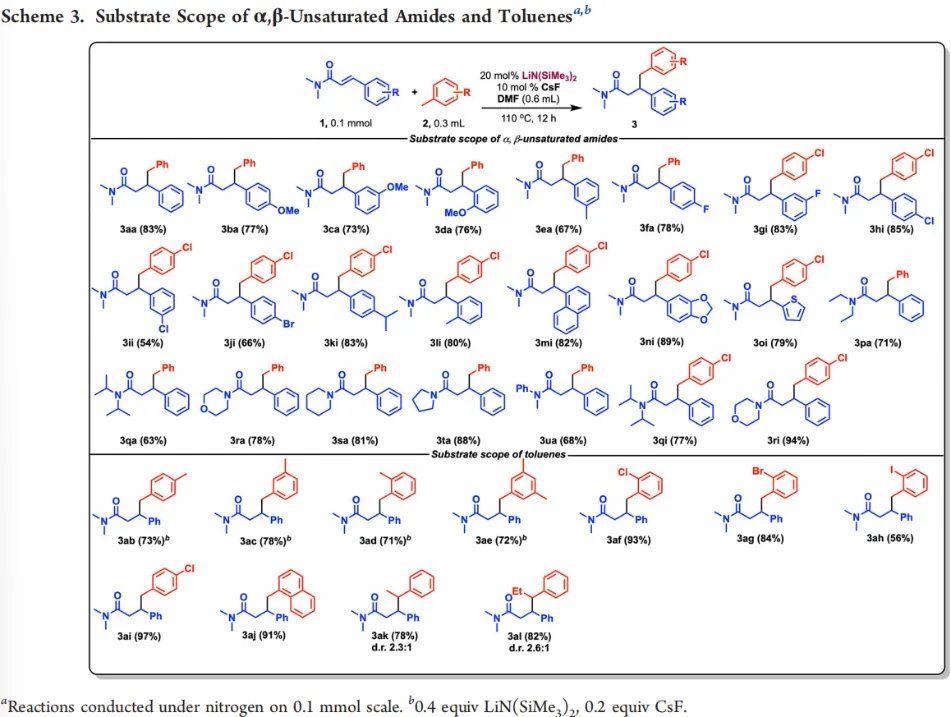
在最优条件下,作者对α,β-不饱和酰胺与甲苯类底物的适用范围进行考察。对于含各类取代基的N,N-二甲基肉桂酰胺,包括含甲氧基、甲基、卤素、萘基与杂环的底物均可顺利反应,以良好至优异收率得到目标产物。甲苯、卤代甲苯、多甲基取代甲苯、乙苯、丙苯、甲基萘等底物均能兼容反应,多甲基取代甲苯需适当提高催化剂用量即可获得理想收率。该体系同样适用于N,N-二乙基、N,N-二异丙基等不同氮取代的α,β-不饱和酰胺,而α,β-不饱和酮、肉桂醛等羰基化合物则难以实现目标转化(Scheme 3)。
(图源:ACS Catal.)
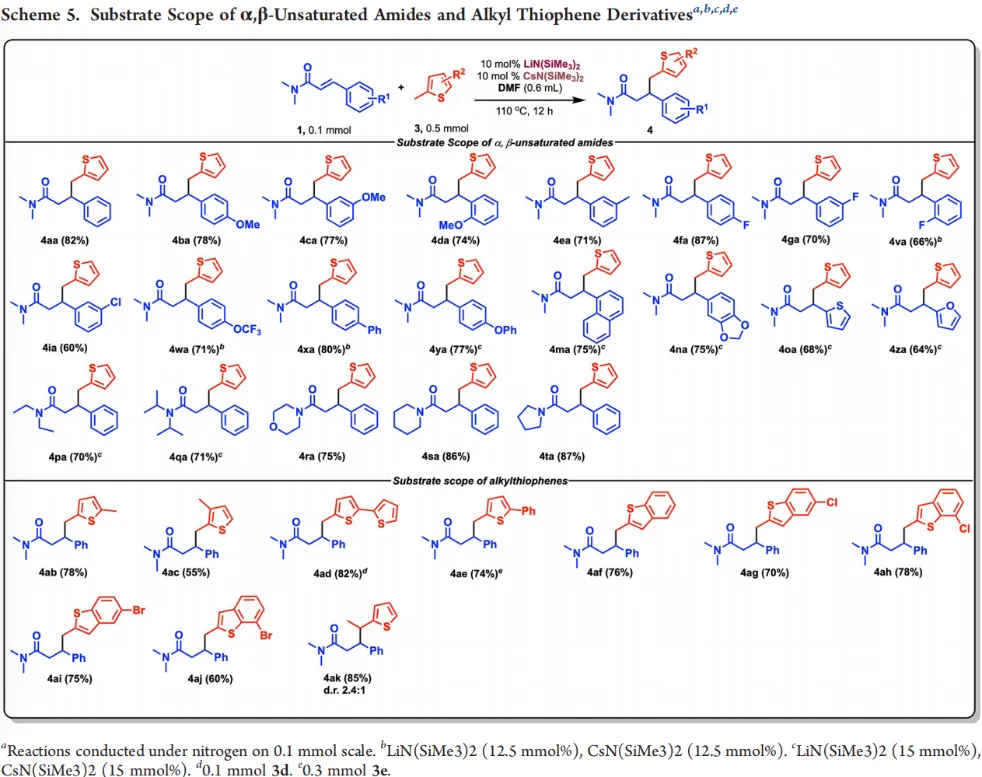
为进一步拓展催化体系的应用,作者将底物范围扩展至2-甲基噻吩类杂环底物(pKa≈42)。经过条件调整,确定以10 mol% LiN(SiMe₃)₂与10 mol% CsN(SiMe₃)₂作为催化剂,可高效实现2-甲基噻吩及其衍生物与α,β-不饱和酰胺的选择性1,4-加成。各类芳基、杂芳基取代α,β-不饱和酰胺均表现出良好反应性,烷基取代、卤素取代的2-甲基噻吩同样适用,以55%–87%收率得到一系列噻吩基取代酰胺衍生物,首次实现烷基噻吩C(sp³)−H键对α,β-不饱和酰胺的催化1,4-加成(Scheme 5)。
(图源:ACS Catal.)
该反应可顺利进行克级规模放大,5 mmol规模下仍能以76%-90%收率得到目标产物,展现出规模化制备潜力。加成产物中的酰胺基团可经水解、还原、酯化等经典转化,高效制备羧酸、胺、酯等重要中间体。其中,TRPV1拮抗剂关键中间体的合成路线由传统5步缩短至3步,总收率由19%-25%提升至29%,在药物分子高效合成中具备显著应用价值(Scheme 6)。
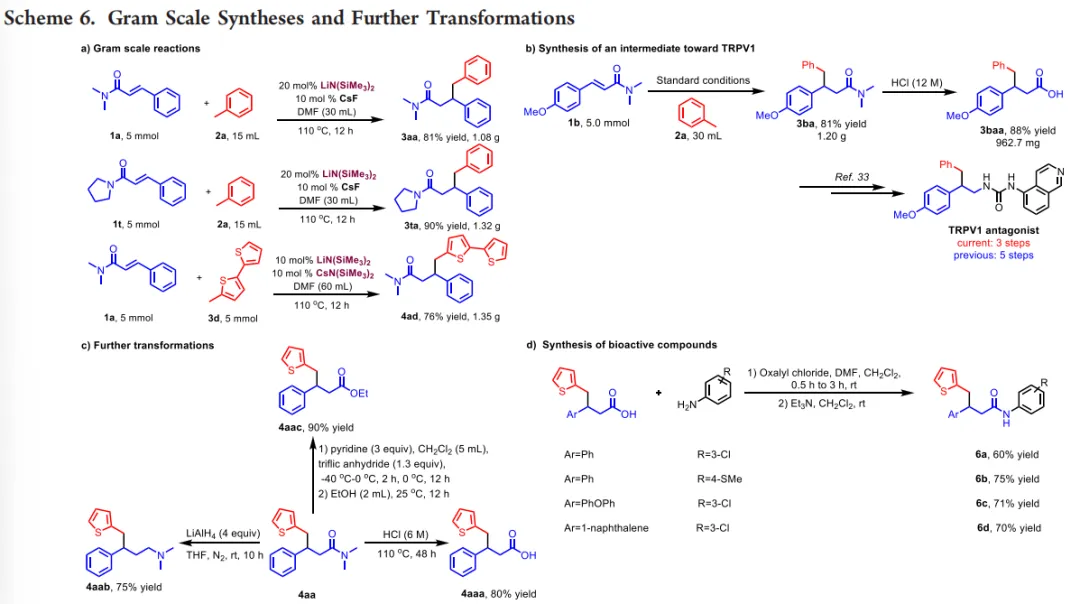
(图源:ACS Catal.)
机理研究表明,反应的平行动力学同位素效应KIE=4.4,证实甲苯苄位C−H键断裂为该催化反应的决速步。对比实验证实,LiN(SiMe₃)₂与CsF原位生成CsLi[N(SiMe₃)₂]₂异双金属活性物种,且氟离子不参与催化循环,仅通过金属交换过程推动活性物种形成。冠醚配位实验显示,封闭Li⁺或Cs⁺的配位位点会显著降低反应效率,证明两种金属的开放配位位点对协同催化至关重要。
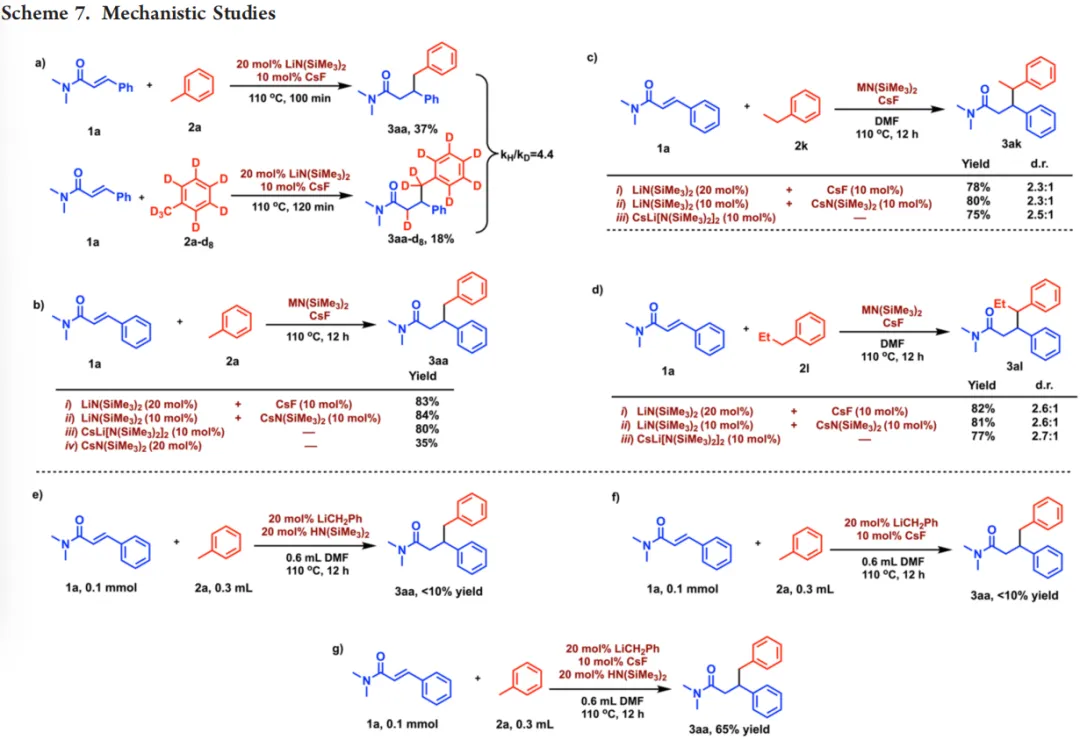
(图源:ACS Catal.)
DFT计算深入揭示了Li⁺/Cs⁺协同催化机制。异双金属Li⁺/Cs⁺组合的去质子化能垒(24.0 kcal/mol)远低于同金属Cs⁺/Cs⁺体系(34.3 kcal/mol),外部π-活化过渡态TS₁-C₂能垒低至19.4 kcal/mol,为最优反应路径。Cs⁺通过强阳离子-π相互作用活化芳环,增强苄位C−H键酸性。Li⁺半径更小,可稳定氮负离子、缩短质子转移距离,提升过渡态稳定性。整个催化循环中,sp³C−H键断裂为决速步,总能垒为21.9 kcal/mol,与实验观测完全吻合。此外,计算得到的反应势能面、理论KIE值均与实验结果高度吻合,直接验证了计算模型与催化机理的可靠性。对氯甲苯等取代底物计算能垒更低,完美解释底物活性差异。
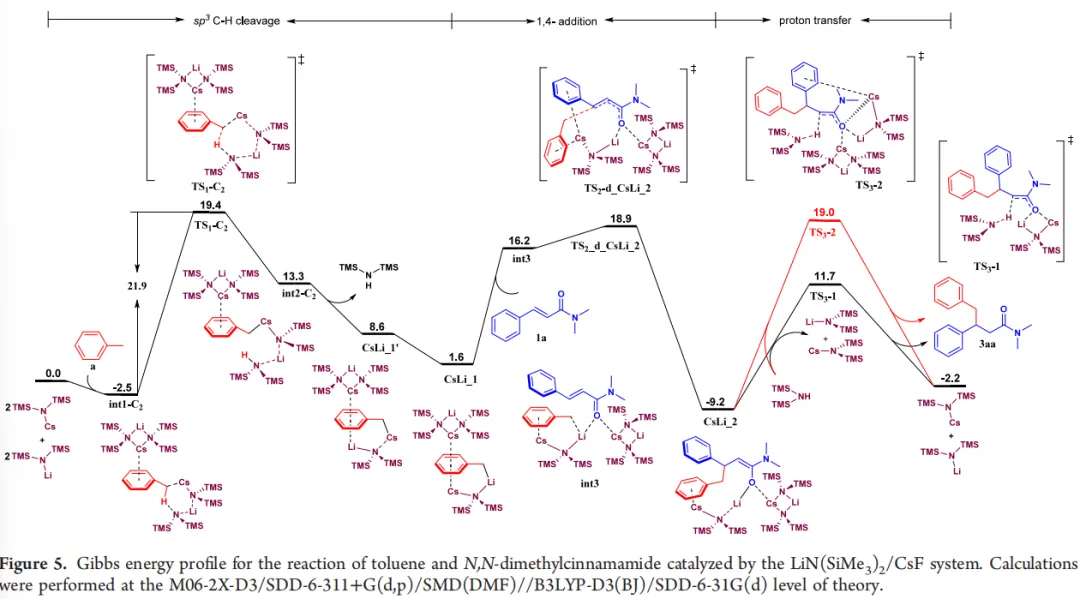
(图源:ACS Catal.)
总结
南京工业大学、南开大学与宾夕法尼亚大学合作团队开发了LiN(SiMe₃)₂/Cs⁺异双金属碱催化体系,成功实现极弱酸性C−H键(pKa>40)对α,β-不饱和酰胺的1,4-加成,将布朗斯特碱催化的1,4-加成反应的前亲核试剂pKa上限由35提升至40以上。反应以甲苯、2-甲基噻吩等大宗化学品作为亲核试剂,无需使用有机金属试剂与过渡金属催化剂,原子经济性高、底物普适性广、可规模化放大。实验与计算结合阐明了Li⁺与Cs⁺协同的阳离子−π活化与去质子化机制,为异碱金属催化体系的设计提供了通用策略,为药物分子、功能材料中C(sp³)−C(sp³)键的绿色构建开辟了新路径。
论文信息
Raising the pKa Limit of Pronucleophiles in 1,4-Additions via Heterobimetallic Alkali-Metal Catalysis.
Yuanyun Gu, Ruyuan Ma, Shenhao Zuo, Guoqing Liu, Guorong Li, Rui Yu, Dan Xiong, Xiufang Xu,* Patrick. J. Walsh,* Jianyou Mao*
ACS Catalysis, DOI: 10.1021/acscatal.6c01195

| 点击即可阅读合集 | ||
| 催化化学 | ||
| 分析化学 | ||
| 生物化学 |

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 南京持续纠治涉企执法领域突出问题,清障破阻为企减负
- 担当引领、协同共进|这场大会,南京民营企业家们这样说……
- 4、5月+五一发班 7星旅聚-南京超五星『悬崖深坑-蜂巢酒店』499元起◆含2早3正◆升级1顿价值88元自助晚餐◆棋牌畅打 纯玩套班三日
- 南京越界梦幻城|藏在新街口的小众文创园!
- 南京威雅关停,不是办学失败,而是理想主义教育,撞上国内极致功利教育生态的必然溃败,道尽二三线城市国际学校的生存真相
- “马什么梅”补货啦!南京半程马拉松官方文创购买→
- 南京工业大学城乡规划考研历年真题初复试全套资料,文末赠送历年真题!
- 【抢先看】南京4月24日启用“南京溧水全民阅读活动周”多色邮资机宣传戳
- 观侵华日军南京大屠杀遇难同胞纪念馆
- 2026/4/22南京融通金报价
