南京大学李朝升等Nature Communications丨d区金属氢溢流效应的起源:从热力学-动力学定量描述到轨道级机制解析
文章标题:
Origins of the hydrogen spillover effect in d-block metals
(d区金属氢溢流效应的起源)
第一作者:
Yang Li, Yuanming Zhang, Zhaojian Zeng(共同第一作者)
通讯作者:
Zhaosheng Li(李朝升)
通讯作者单位:
- 南京大学工程与应用科学学院,固体微结构国家重点实验室,先进微结构协同创新中心
- 南京大学江苏省纳米技术重点实验室
论文DOI:
10.1038/s41467-026-72608-0
期刊:
Nature Communications, 2026
研究背景:氢溢流——催化界的“百年谜题”
氢溢流(hydrogen spillover)是指H₂在活性金属(如Pt、Pd、Ru)表面解离吸附后,生成的氢原子自发迁移到原本难以活化H₂的载体表面上的现象。自1964年Khoobiar首次发现以来,氢溢流已被证实广泛存在于加氢、氢解、氢储存、CO₂还原等众多催化过程中,是理解多相催化反应机理的关键。
然而,尽管经过了半个多世纪的研究,氢溢流效应的定量描述和普适性机制仍然缺失。传统研究主要面临三大挑战:
- 活化区与溢流区难以解耦:H₂在金属上解离(活化)和H原子在载体上迁移(溢流)同时发生,信号重叠,无法区分两者的独立贡献。
- 缺乏定量描述符:目前对氢溢流的评价多停留在“有无”或“强弱”的定性层面,没有统一的量化指标来比较不同金属的溢流能力。
- 机制不统一:不同金属(如Ru、Pd、Ni)表现出截然不同的溢流行为,但其根源(是电子结构?是d带中心?还是d-s轨道杂化?)尚不清晰。
本文的核心科学问题:能否发展一套定量方法,解耦H₂活化与氢原子溢流,并揭示d区金属氢溢流能力的电子结构起源?这一问题不仅对理解催化机理至关重要,更直接关系到高效加氢催化剂和化学储氢材料的理性设计。
全文速览:从“定性描述”到“定量图谱”——氢溢流研究的范式转变
本文首次提出了一套完整的热力学-动力学定量描述框架,通过光激发还原(PER)测试,将氢溢流过程成功解耦为“活化区”(快速H₂解离)和“溢流区”(慢速H原子迁移),并定义了四个关键描述符:
- 溢流容量(Γ):单位质量催化剂活化H₂的总量。
- 比溢流容量(Γ*):单位质量金属活化H₂的量(反映金属本征活性)。
- 活化度(Φ):峰值活化速率(反映H₂解离动力学)。
- 平均溢流速率(κ):溢流区的平均迁移速率(反映H原子在载体上的扩散行为)。
利用该平台,作者系统研究了负载在还原性TiO₂₋ₓ上的五种d区金属(Mn、Fe、Co、Ni、Ru)。结果显示,Ru在所有金属中展现出最高的Γ、Γ*、Φ和κ,而其溢流能力与金属的d-s轨道杂化程度呈非线性正相关。
通过DFT计算、AIMD模拟和XAS表征,作者进一步揭示了电子结构层面的机制:d区金属中部分空位的d-s轨道与吸附H的σ和σ*轨道发生协同重叠,形成弱反键态,从而同时削弱H-H键(促进解离)和M-H键(促进迁移)。Ru因其独特的4d⁷5s¹电子构型,d-s杂化最强,因此表现出最优异的氢溢流性能。
作为应用验证,利用该定量平台筛选出的最优Ru/TiO₂₋ₓ催化剂在光驱动CO₂加氢反应中实现了948 mmol·g⁻¹·h⁻¹的CH₄产率,创下新纪录,并稳定运行100小时。
文章亮点
⭐提出氢溢流定量描述框架:首次通过光激发还原测试将活化区与溢流区解耦,定义了Γ、Γ、Φ、κ四个量化描述符,为氢溢流研究提供了统一标尺。⭐揭示d-s轨道杂化是溢流能力的电子起源:理论计算表明,部分空位的d-s轨道与H的σ/σ轨道协同重叠形成弱反键态,同时削弱H-H和M-H键,这是非线性溢流能力的内在原因。⭐Ru的卓越性能源于其d-s杂化优势:Ru(4d⁷5s¹)的d-s杂化程度最高,Γₘₐₓ达523.3 mmol·gRu⁻¹,Φ和κ均远高于其他金属。⭐定量平台指导催化剂设计:利用Γ识别最优金属负载量(避免活化区重叠),成功优化出高效光热CO₂加氢催化剂,CH₄产率948 mmol·g⁻¹·h⁻¹,100小时稳定。⭐实验与理论高度吻合:PER测试、AIMD模拟、DFT计算和XAS/XPS表征结果自洽,完整建立了“宏观性能→微观动力学→电子结构”的证据链。
图文解析
图1. 氢溢流定量描述平台的定义与Ru/TiO₂₋ₓ的验证
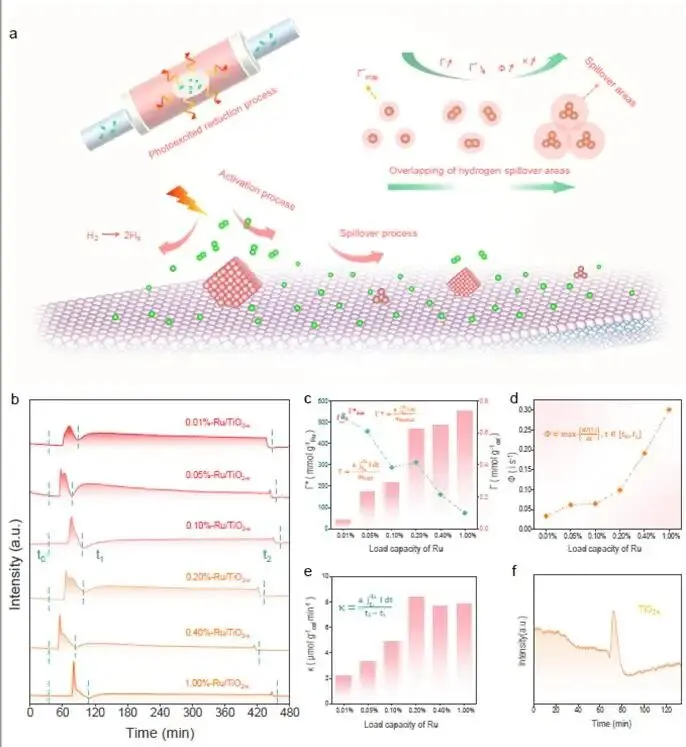
图1a | 氢溢流过程示意图将氢溢流分为两个时序过程:
- 活化区(t₀ → t₁):H₂在金属表面快速解离为H原子(Hs)。
- 溢流区(t₁ → t₂):Hs从金属表面迁移到载体表面(扩散控制,较慢)。定义了四个描述符:
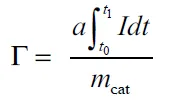 (总溢流容量)
(总溢流容量)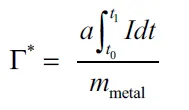 (比溢流容量,反映金属本征活性)
(比溢流容量,反映金属本征活性)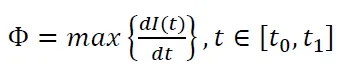 (活化度,峰值活化速率)
(活化度,峰值活化速率)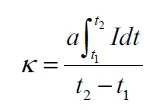 (平均溢流速率)
(平均溢流速率)
图1b | 不同Ru负载量的PER测试曲线
- 横轴为时间,纵轴为检测信号(与H₂消耗量成正比)。
- 每个样品均出现尖锐的活化峰(t₀-t₁)和宽缓的溢流峰(t₁-t₂),证实了两者可解耦。
- 随着Ru负载量从0.01%增至1.00%,活化峰和溢流峰的强度均显著增加。
图1c | Γ和Γ*随Ru负载量的变化
- Γ(总容量)随Ru负载量增加而单调增加(更多金属,更多H₂活化)。
- Γ(比容量)随负载量增加先保持不变后下降:在低负载(<0.10%)时,Ru原子孤立分散,各活化区不重叠,Γ达到最大值523.3 mmol·g_Ru⁻¹;当负载超过0.20%时,Ru原子间距缩小,活化区重叠,单位Ru的活化效率下降。
- 这一发现明确了孤立分散是最大化金属原子利用效率的关键。
图1d | Φ(活化度)随Ru负载量的变化
- Φ随Ru负载量增加而单调增加(更多Ru位点,更快达到峰值活化速率)。
图1e | κ(平均溢流速率)随Ru负载量的变化
- κ在低负载时随Ru负载量增加而快速上升,在~0.20%后趋于饱和。
- 表明溢流速率受限于载体表面的扩散通道,一旦金属覆盖度达到临界值,再增加金属对溢流速率贡献不大。
图1f | 纯TiO₂₋ₓ的PER测试
- 几乎没有活化峰和溢流峰,证实纯载体本身无法活化H₂。
科学意义:图1首次实现了氢溢流过程的定量解耦,并揭示了负载量对Γ*的非线性影响(孤立分散→最优比容量→团聚/重叠→比容量下降)。这为理性设计低贵金属负载量催化剂提供了直接依据。
图2. d区金属氢溢流能力的系统对比与轨道级机制
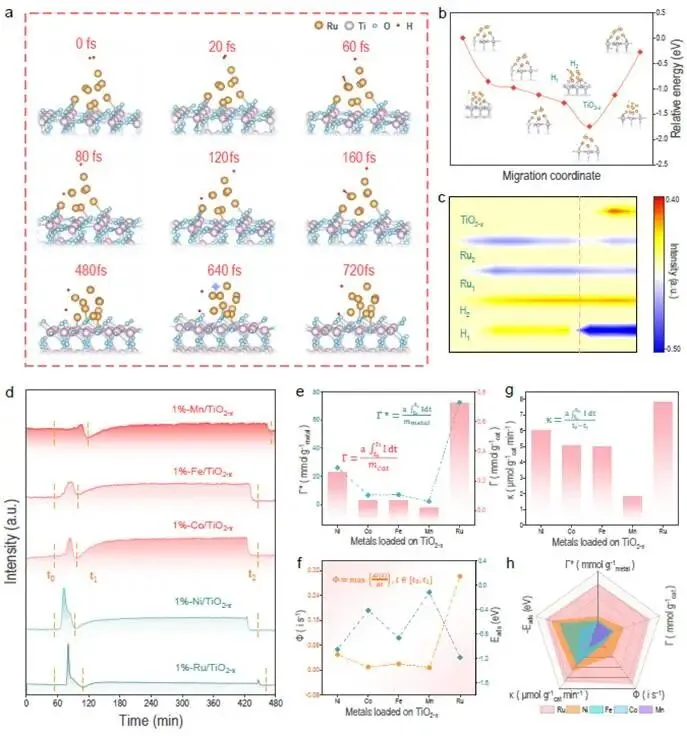
图2a-c | AIMD模拟的氢溢流过程(Ru/TiO₂₋ₓ)
- 0 fs:H₂分子靠近Ru簇。
- 20-60 fs:H₂在Ru表面解离,形成Ru-H键(活化区)。
- 80-160 fs:一个H原子(标记为H₂)留在Ru上,另一个H原子(H₁)跨越Ru/TiO₂界面扩散到载体上(溢流区)。
- 480-720 fs:H₁在TiO₂₋ₓ表面形成Ti-O-H物种,H₂继续留在Ru上。
- 图2c显示了电荷转移过程:从Ru到H(活化)和从H到载体(溢流)。
图2d | 五种金属(Mn、Fe、Co、Ni、Ru)的PER曲线
- 所有样品均清晰显示活化峰和溢流峰。
- 活化峰强度和溢流峰面积:Ru > Ni > Co > Fe > Mn。
图2e | Γ和Γ*的对比
- Γ(总容量)顺序:Ru > Ni > Co > Fe > Mn。
- Γ*(比容量)顺序:Ru > Ni > Co > Fe > Mn,与H₂吸附能(Eads)的绝对值正相关(图2h)。
图2f | Φ(活化度)的对比
- 顺序:Ru > Ni > Co > Fe > Mn,同样与|Eads|正相关。
图2g | κ(平均溢流速率)的对比
- 顺序:Ru > Ni > Co > Fe > Mn,与d-s轨道杂化程度正相关。
- 从表2a可见,Mn(3d⁵4s²)→ Fe(3d⁶4s²)→ Co(3d⁷4s²)→ Ni(3d⁸4s²)→ Ru(4d⁷5s¹),d-s杂化逐渐增强,κ值单调增加。
图2h | 与DFT计算E_ads的对比
- 插图显示,Γ*、Φ与|E_ads|呈正相关。
- Ru的E_ads最负(~-2.2 eV),因此活化能力最强。
科学意义:图2通过AIMD直观展示了氢溢流的原子过程,并用定量描述符系统比较了五种金属的溢流能力。Ru在活化与溢流两方面均表现最优,揭示了d-s轨道杂化程度是溢流能力的关键描述符。
图3. Ru/TiO₂₋ₓ的结构与化学异质性
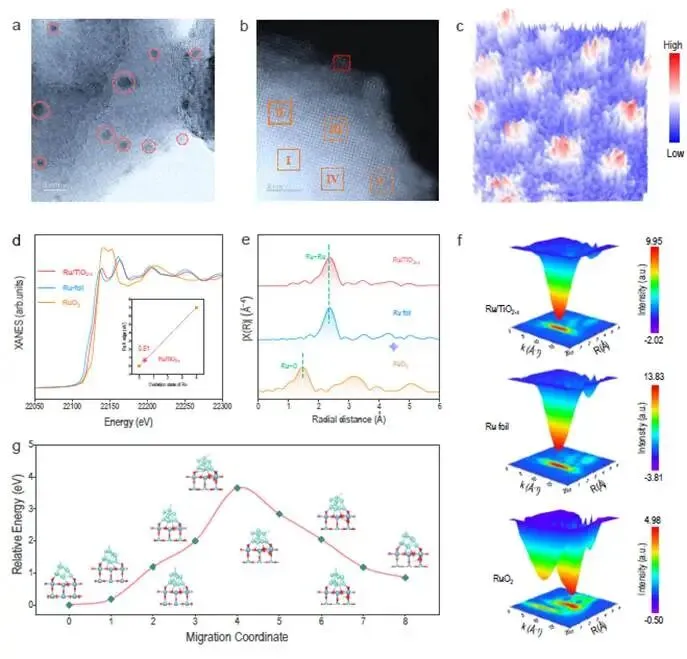
图3a-b | HAADF-STEM图像
- Ru纳米簇(~1-2 nm)均匀锚定在TiO₂₋ₓ表面,形成清晰的界面(图3b)。
- 图3c显示三维强度等高线,Ru簇呈现有序原子排布,无团聚。
图3d | Ru K-edge XANES
- Ru/TiO₂₋ₓ的吸收边位于Ru foil和RuO₂之间,更接近Ru foil。
- 线性拟合得平均氧化态≈0.52,表明大多数Ru为金属态(Ru⁰),少量Ru-O界面作用。
图3e | FT-EXAFS(R空间)
- Ru/TiO₂₋ₓ在~2.45 Å出现Ru-Ru配位峰(金属Ru),无明显的Ru-O峰(~1.6 Å),证实金属Ru簇为主相。
- 对比Ru foil(更尖锐)和RuO₂(~1.9 Å的Ru-O峰),差异明显。
图3f | WT-EXAFS(k空间)
- Ru/TiO₂₋ₓ在k~8 Å⁻¹、R~2.5 Å处出现单一高强度峰,对应Ru-Ru配位。
- 没有Ru-O或Ru-载体其他配位的特征峰。
图3g | H原子迁移进入TiO₂₋ₓ体相的吉布斯自由能变化
- 计算表明,H从表面迁移进入体相的能垒非常高(>2 eV),因此氢溢流主要发生在表面和界面,而非体相扩散。
科学意义:图3确认了Ru/TiO₂₋ₓ中Ru以金属态纳米簇形式存在,与载体形成清晰界面,且H原子难以进入TiO₂体相——这解释了氢溢流被限制在表面/界面的结构原因。
图4. 定量氢溢流在CO₂加氢中的应用
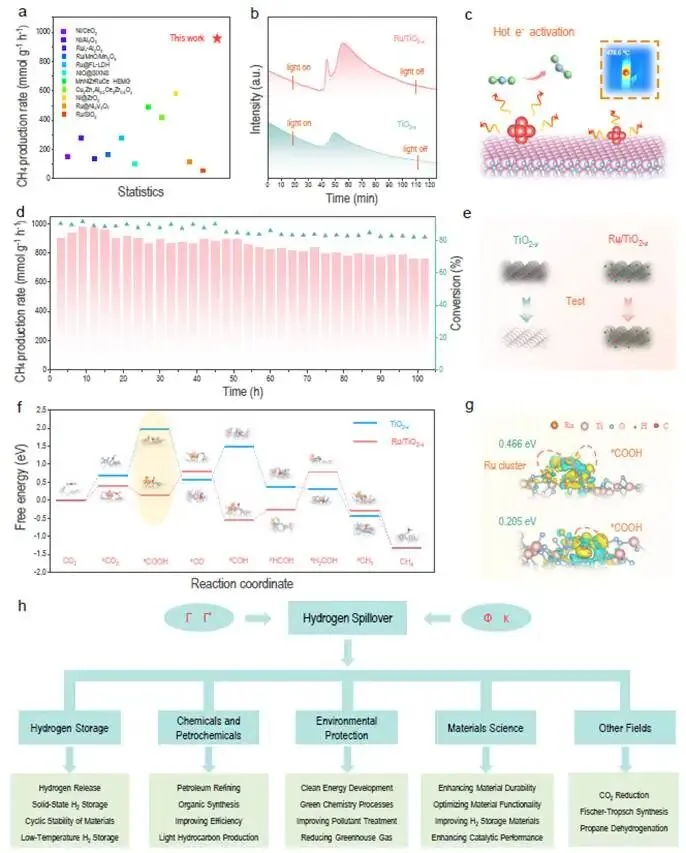
图4a | 光热催化CO₂加氢性能对比
- Ru/TiO₂₋ₓ的CH₄产率达948 mmol·g⁻¹·h⁻¹,远超已报道的光驱动和热催化Sabatier反应催化剂。
- 补充表2列出了详细对比数据。
图4b | 光激发CO₂脱附测试
- 光照诱导CO₂从催化剂表面脱附,信号强度反映活化CO₂的数量(光活性位点密度)。
- Ru/TiO₂₋ₓ的脱附峰面积远大于纯TiO₂₋ₓ,证明Ru簇促进了CO₂的活化。
图4c | 热电子注入活化CO₂示意图
- 光激发下,Ru簇产生热电子,注入到吸附的CO₂的π*反键轨道,削弱C=O键,促进活化。
图4d | 100小时稳定性测试
- Ru/TiO₂₋ₓ在连续100小时光照下,CH₄产率和选择性无明显衰减。
- 相比之下,纯TiO₂₋ₓ快速失活。
图4e | 催化剂颜色演变
- 反应前:黑色(氧空位和Ti³⁺缺陷)。
- 反应后:纯TiO₂₋ₓ变白(缺陷被氧化修复),而Ru/TiO₂₋ₓ仍保持黑色。
- Ru通过氢溢流持续提供电子,抑制缺陷氧化,维持活性。
图4f | DFT计算的C₁路径自由能图
- *CO₂ → *COOH步骤:在纯TiO₂₋ₓ上吸热3.18 eV,在Ru/TiO₂₋ₓ上放热1.23 eV。
- 表明Ru簇极大促进了CO₂的活化。
*图4g | COOH中间体的差分电荷密度
- Ru/TiO₂₋ₓ模型中,电荷从Ru/TiO₂转移到*COOH更多,键合更强。
图4h | 定量氢溢流的应用前景示意图
- 提出的描述符可应用于:光热催化、化学储氢、丙烷脱氢等。
科学意义:图4验证了定量氢溢流平台指导催化剂设计的有效性——利用Γ*识别最优Ru负载,结合氢溢流增强的电子供应,构建了高效、稳定的CO₂加氢催化剂。
总结与展望
本研究通过发展一套原创的氢溢流定量描述框架,成功解耦了H₂活化与氢原子溢流,首次系统比较了d区金属(Mn、Fe、Co、Ni、Ru)的溢流能力,并揭示了其电子结构起源——d-s轨道杂化程度。主要科学突破包括:
- 定量描述符体系:定义了Γ、Γ*、Φ、κ四个量化指标,实现了氢溢流从“定性”到“定量”的跨越。
- 解耦活化与溢流:光激发还原测试(PER)克服了传统方法信号重叠的难题,首次清晰区分两个过程。
- 机制揭示:DFT加AIMD表明,d区金属部分空位的d-s轨道与H的σ/σ*轨道协同重叠形成弱反键态,同时削弱H-H键(促进解离)和M-H键(促进迁移)。
- Ru的最佳表现:Ru(4d⁷5s¹)具有最强的d-s杂化,Γ*ₘₐₓ达523.3 mmol·g_Ru⁻¹,Φ和κ均最高。
- 应用验证:利用Γ*筛选最优Ru负载(孤立分散),构建的Ru/TiO₂₋ₓ催化剂在光热CO₂加氢中实现创纪录的CH₄产率(948 mmol·g⁻¹·h⁻¹)和100小时稳定性。
未来展望:
- 拓展金属种类:将定量平台应用于更多d区金属(Pd、Pt、Ir)以及非d区金属(如Cu、Ag),验证普适性。
- 载体效应研究:系统比较不同载体(Al₂O₃、CeO₂、ZrO₂等)对κ的影响,建立载体-溢流速率构效关系。
- 单原子催化剂:利用Γ*识别孤立单原子的最大本征活性,指导单原子催化剂的理性负载量设计。
- 工业应用:将定量平台集成到自动化催化剂筛选系统中,加速加氢、储氢、脱氢等领域的催化剂开发。
通讯作者简介
李朝升(Zhaosheng Li):1975年出生。2003年获得中国科学院研究生院理学博士学位;2003年至2005年在南京大学环境材料与再生能源研究中心从事博士后研究工作(其间2004年2-3月在日本国立材料研究所做访问学者); 2005年11月起在南京大学材料科学与工程系和环境材料与再生能源研究中心工作。2005年11月起在南京大学材料系担任讲师;2006年12月晋升副教授;2007年6-8月在日本国家材料研究所ICYS做访问研究;2011年12月晋升教授。现为国家杰出青年基金获得者、江苏特聘教授、国家基金委会评专家。在Nat. Mater.、Nat. Sustain.、Joule、NSR、PNAS、JACS、Angew. Chem.等期刊发表学术论文多篇,被引用2.2万余次。授权发明专利21件。获江苏省科学技术一等奖2项(第一完成人和第二完成人各1项)、国家自然科学二等奖1项(第二完成人)。
声明:
文章内容仅代表本人观点,本文数据来源于Li et al. Nature Communications, 2026, DOI: 10.1038/s41467-026-72608-0论文,图片版权归原作者和出版社所有。如有侵权,请联系后台,感谢支持,欢迎批评指正!
欢迎各位老师同学们在公众号上投稿分享课题组研究成果,不限期刊和发表时间!后台联系即可。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
