
活体组织会持续重塑其结构以适应不断变化的力学环境,在频繁使用中增强自身强度的同时保持柔韧性和恢复能力。值得注意的是,这种适应机制不仅限于变形过程中的可逆应变硬化,更包含由力学敏感生化反馈驱动的不可逆基质重塑。在合成软材料中模拟这种自我强化特性一直是长期追求的目标,但由于将机械应力与可控的分子定向排列或化学键生成相耦合存在本质困难,这一目标至今仍难以实现。活体组织在反复机械加载下会增强自身强度,但在合成材料中复现这种适应性生长仍是重大挑战。
来自南京大学的王炜等团队报道了一种基于蛋白质的水凝胶,其能通过机械化学诱导的自生长机制,在外加应力作用下自主强化基础力学性能。该策略利用铜储存蛋白Csp1:力调控的蛋白去折叠过程会释放Cu(I),进而催化原位叠氮-炔烃环加成反应,在机械载荷下生成次级交联键。卸载后,Csp1重新折叠并再捕获Cu(I),从而终止催化反应并恢复生长能力。这种机械催化反馈回路使封闭系统(无需外部单体供应)内实现应力与时间依赖的自强化过程。通过利用循环生长-暂停-生长转变中的Cu(I)稳态,该水凝胶可展现可编程的力学记忆特性。本研究通过将力依赖的蛋白质构象动态变化与催化活性相耦合,为设计结构与功能随力学刺激演化的自适应生物材料建立了通用型机械化学框架。相关工作以题为“Mechanically Induced Adaptive Self-Growing Protein Hydrogel”的文章发表在2026年05月02日的期刊《Advanced Materials》。

【机械诱导自适应自生长蛋白水凝胶的设计】
在经典的自生长水凝胶中,机械强化源于化学键断裂产生的自由基,这些自由基会引发新网络的聚合反应。在此过程中,初始网络的断裂首先会削弱材料,但随后通过形成次级交联键/网络来增强其强度(图1a)。然而,这种强化过程一旦启动便无法停止,因为聚合反应会持续进行直至封闭系统中的所有可用单体被消耗殆尽。因此,这种行为对应的是机械触发型而非机械调控型生长。为实现可控的自生长行为,本文引入了一个机械响应型蛋白质去折叠-重折叠机制来调控催化离子的释放与重捕获,从而调节离子催化的交联反应(图1b)。由此,水凝胶在机械训练过程中持续增强,但一旦形变释放便迅速停止,实现了阶梯式的机械调控自生长过程。与无需等待时间即可在连续拉伸-松弛循环中进行的键断裂触发型自生长(图1a插图)不同,离子释放诱导型自生长需要在最大或最小应变处保持一定时间。这种维持时间可使释放的离子浓度保持在临界催化阈值以上(图1b插图)。此外,由于可逆蛋白质去折叠-重折叠的快速动力学特性,释放的离子可被蛋白质迅速重新捕获,从而在蛋白质水凝胶中实现机械可控的自生长启动与暂停。
为实现上述构想,本文设计了一种水凝胶体系:以铜储存蛋白Csp1作为主要交联剂,聚丙烯酰胺(PAAm)构成渗透相,并将Cu(I)催化功能基团(叠氮基和炔基)预先整合至网络中。Csp1是一种从甲基弯菌OB3b中鉴定出的蛋白质,因其通过富含半胱氨酸的四螺旋束结构可结合多达13个Cu(I)离子而被选为模型组分。在拉伸形变下,Csp1发生去折叠并释放Cu(I)离子,这些离子随即启动Cu(I)催化的叠氮-炔烃环加成反应(CuAAC),生成次级交联键,从而驱动水凝胶的机械自生长(图1c)。松弛状态下,Csp1重新折叠并重新捕获Cu(I)离子,导致CuAAC反应和机械生长立即终止。当再次施加形变时,该过程重新启动,实现适应性、阶梯式的自生长行为(图1c右图)。值得注意的是,由于叠氮基和炔基前体已预先载入水凝胶网络,该系统在无需外部单体供应的封闭环境中运行。
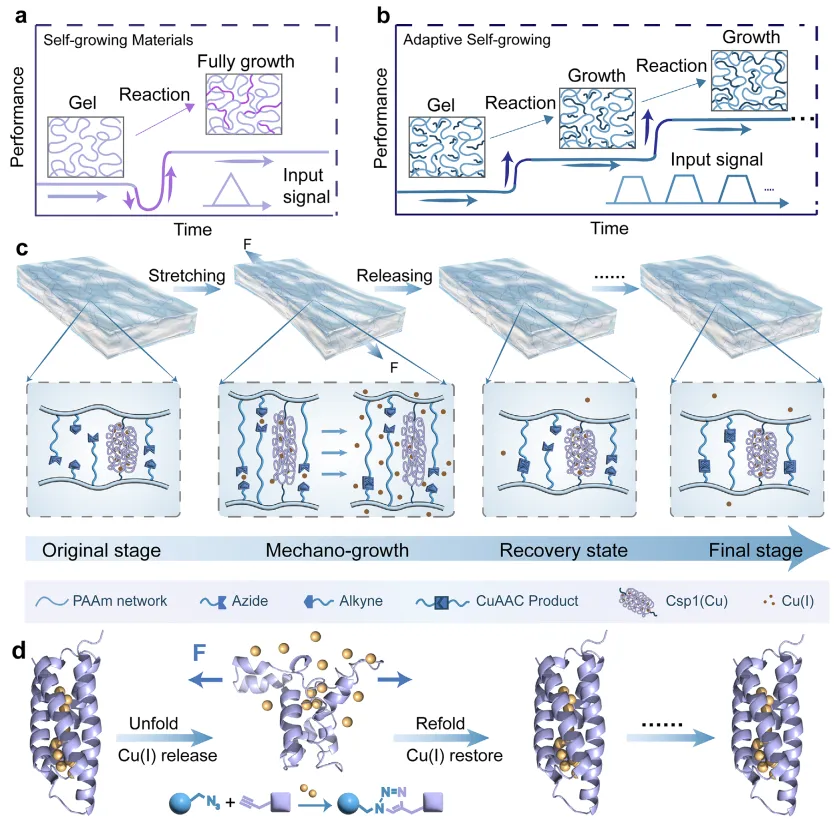
图1 机械诱导自适应自生长蛋白水凝胶的设计
【通过蛋白质去折叠-再折叠实现Cu(I)的可逆释放与恢复】
为验证铜储存蛋白Csp1是否能在力的作用下可逆地释放和重新摄取Cu(I)离子,本文首先利用基于原子力显微镜(AFM)的力谱技术,在单分子水平上研究了其机械去折叠行为(图2a)。为此,本文构建了一种嵌合多聚蛋白Fgβ-(GB1)₂-Csp1-(GB1)₂-SpyTag,用于单分子力谱(SMFS)测量。该构建体通过SpyCatcher/SpyTag自发连接反应,共价锚定在经cys-GB1-SpyCatcher修饰的基底上,而其N端的Fgβ结构域则被SdrG修饰的AFM悬臂特异性识别并拾取。拉伸过程中,获得了锯齿状的力-延伸曲线,其中Csp1的特征性去折叠事件出现在约120 pN处,伴随约42 nm的轮廓长度增量,而约18 nm ΔLc的峰则对应GB1标记结构域的去折叠(图2b-d)。重要的是,通过对比无Cu(I)的Csp1(apo-Csp1)的SMFS结果证实,铜负载并未显著影响去折叠力或轮廓长度增量。圆二色光谱(CD)进一步表明,Cu(I)结合未引起Csp1二级结构的可检测变化。综合来看,这些结果表明Csp1在金属负载状态下仍保持其结构完整性,而约120 pN的内应力足以诱导Csp1去折叠并释放其所储存的Cu(I)离子。
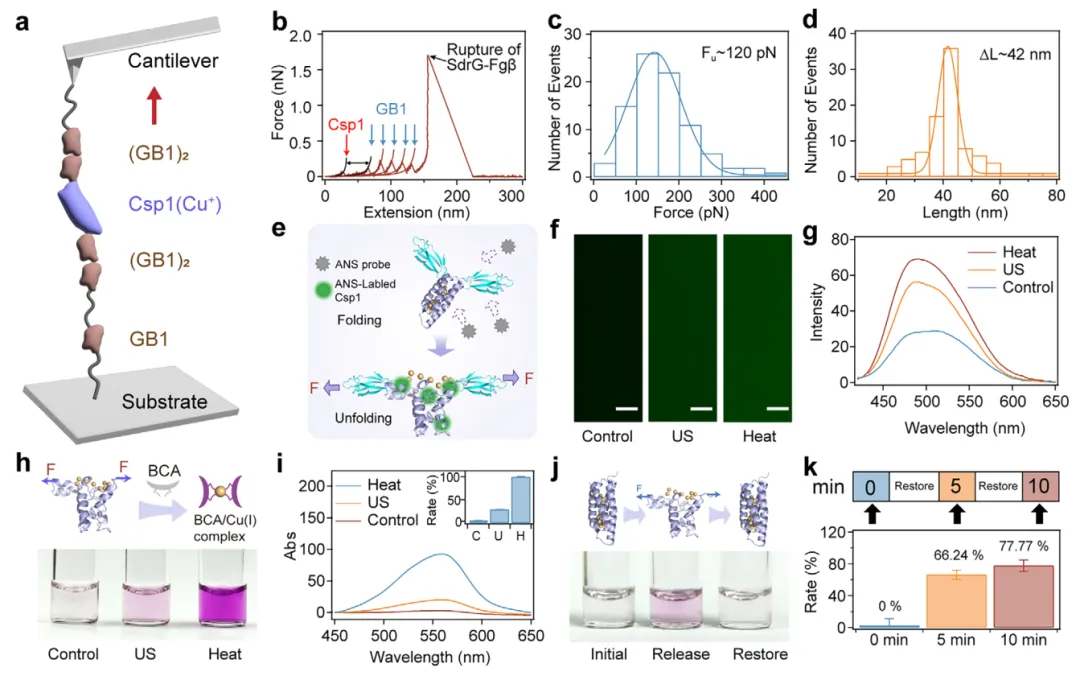
图2 蛋白解折叠介导的Cu(I)释放与重折叠介导的恢复的分子表征
【水凝胶中机械诱导Cu(I)离子的释放与恢复】
为探究水凝胶中机械诱导的Cu(I)释放,本文首先构建了以Csp1作为共价交联剂的单网络水凝胶(图3a)。在HEPES缓冲液中制备了含有丙烯酰胺、丙烯酸酯末端修饰的SpyTag(SpyTag-AA)和SC-Csp1-SC的前驱体溶液,并使用苯基-2,4,6-三甲基苯甲酰基次膦酸锂(LAP)作为光引发剂,在紫外光照射下进行聚合。聚合后通过透析去除未反应的单体,所得水凝胶称为PAAm/Csp1。在该设计中,SC-Csp1-SC通过与SpyTag-AA反应末端功能化修饰上丙烯酸酯基团,使得在共聚过程中Csp1能够作为共价交联剂嵌入到PAAm网络中。本文首先评估了不同Csp1浓度下PAAm/Csp1水凝胶的力学性能。这些水凝胶的断裂应变超过647%,断裂应力高达173 kPa。在拉伸-松弛循环过程中出现了明显的滞后现象,这表明通过力诱导的Csp1去折叠实现了能量耗散。增加应变幅度产生了逐渐增大的滞后面积,证实了蛋白质去折叠与应变的依赖性。含有3 mM Csp1的水凝胶表现出最大的滞后,意味着其去折叠程度最高,因此具有最大的Cu(I)释放潜力。
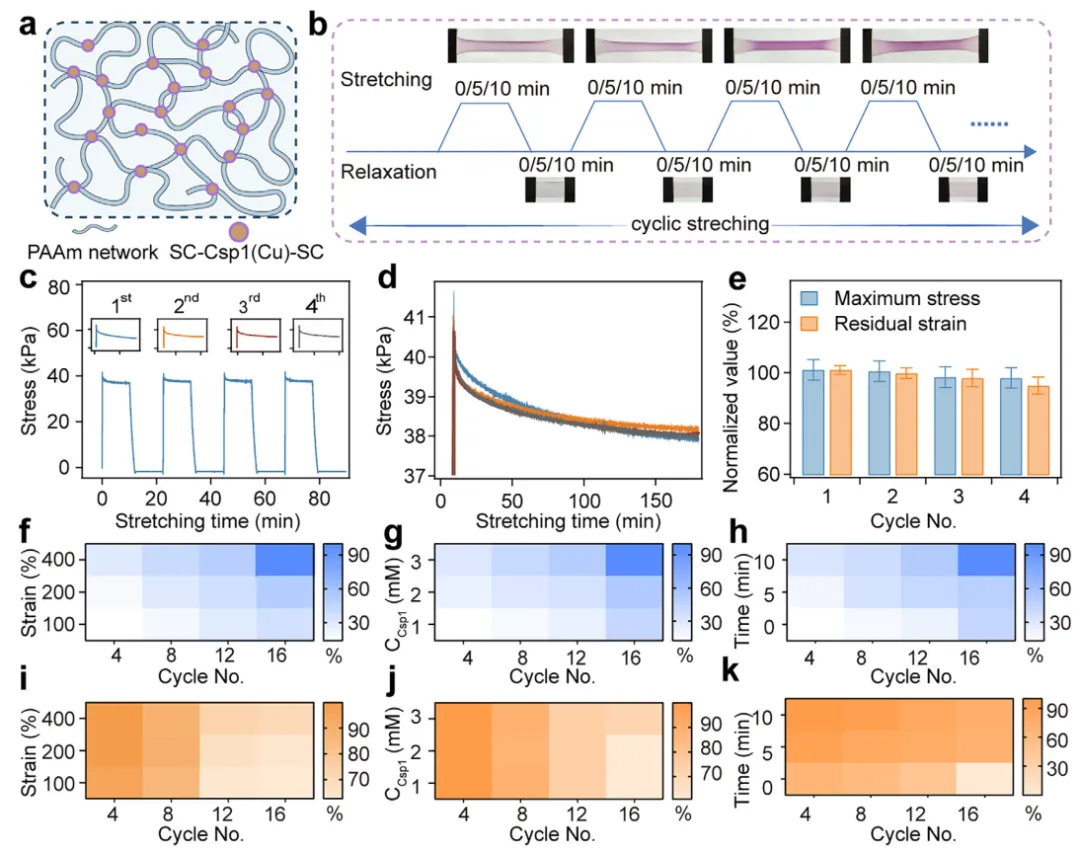
图3 机械诱导Csp1交联水凝胶中Cu(I)离子的释放与恢复
【水凝胶的机械诱导自适应自生长】
为了赋予水凝胶机械自适应生长能力,本文将叠氮基团和炔基团整合到Csp1交联的网络中,以便在机械加载过程中实现Cu(I)催化的叠氮-炔基环加成反应(CuAAC)。所得水凝胶(记为PAAm/Csp1/AA)含有等摩尔比的叠氮和炔基,并接受了循环拉伸-松弛处理(约400%应变;无松弛时间)(图4a)。在最初的拉伸-松弛循环中,杨氏模量和最大应力均迅速增加,到第16次循环时分别提高了超过400%和250%(图4b)。这种渐进的增强源于由机械释放的Cu(I)催化原位CuAAC反应而累积形成的共价交联。超过16次循环后,额外的加载没有产生进一步的显著增强,表明机械生长趋近于一个有限的上限(图4b)。这种饱和主要受限于反应性叠氮/炔基团的消耗,以及随着交联密度增加,剩余功能基团可及性的降低。值得注意的是,Cu(I)的释放和再结合在50次循环中保持稳定,没有出现渐进性损失或失控积累的迹象。相比之下,脱辅基Csp1对照水凝胶(缺乏Cu(I))没有表现出增强,反而在相同的循环条件下显示出轻微的力学性能下降(图4b)。此外,延长的停顿期对照(16次循环后1-7天)没有显示出背景增强,证实了残留的Cu(I)不会积累到足以削弱力门控催化精度的水平。总之,这些结果表明,Csp1介导的机械化学系统作为一个稳健的循环催化模块,确保了金属稳态和机械编码记忆的长期稳定性。
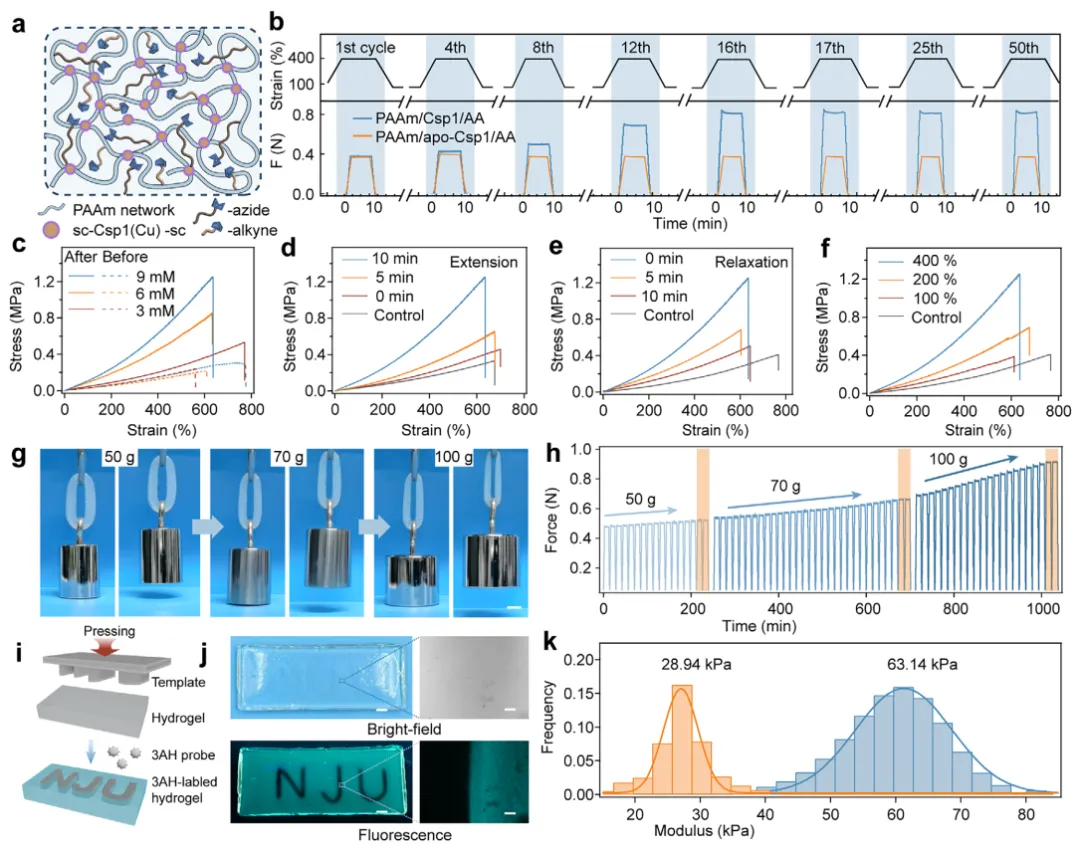
图4 机械触发的时空自生长水凝胶
【通过机械诱导CuAAC反应实现自生长的机理认识】
机械触发的CuAAC反应及其在水凝胶网络中形成二次交联的过程得到了系统验证。如图5a所示,将从不同拉伸-松弛循环次数后的PAAm/Csp1水凝胶中获得的浸出液与3AH混合后,荧光强度随循环次数逐渐增加,表明变形过程中释放的Cu(I)离子具有催化活性,并成功启动了CuAAC反应(图5b,c)。同时,利用3AH对PAAm/Csp1/AA水凝胶内部原位CuAAC反应的效率进行了定量分析(图5d)。将经过递增拉伸-松弛循环次数的水凝胶浸入3AH溶液中1小时,随后测量其荧光强度。随着循环次数增加,荧光强度逐渐下降(图5e),这反映了炔基基团的消耗。定量结果表明,在8次和16次循环后,炔基的消耗率分别约为47.96%和96.47%(图5f),证实了预载的叠氮/炔基对在持续机械激活过程中几乎完全转化。总的来说,交联密度的增加、CuAAC反应效率以及机械模量的增强三者共同受拉伸-松弛诱导自生长过程中施加的应变幅度、循环次数、拉伸持续时间和松弛时间的调控,而机械诱导强化的最终幅度则主要由叠氮/炔基的负载密度决定。
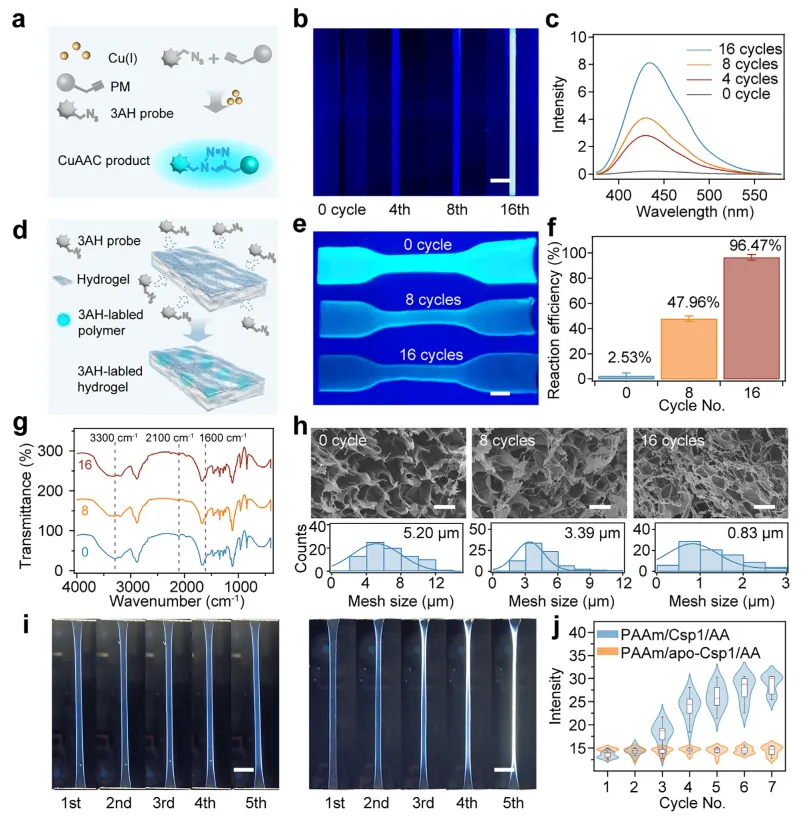
图5 机械触发Cu(I)催化的叠氮-炔基环加成反应(CuAAC)及二次交联形成的机理验证
【总结与展望】
总之,本研究报道的蛋白质介导的闭环机械催化策略定义了一类新型的力学智能材料,弥合了生物力传导与合成力化学之间的鸿沟。利用金属蛋白的可逆解折叠作为催化调控因子,为单网络自演化水凝胶提供了强有力的框架,使其能够在机械刺激下自主优化自身结构。除了基础科学意义外,这一机制在自适应生物材料、软体机器人和植入式系统中具有广阔应用前景,在这些系统中,机械应力同时充当结构演化的信号、催化剂和调控器。
参考资料:
https://doi.org/10.1002/adma.202523636
来源:EngineeringForLife
声明:仅代表作者个人观点,用于研究用途,作者水平有限,如有不科学之处,请在下方留言指正!

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
