电化学合成利用电子作为无痕氧化还原试剂,在温和条件下驱动化学转化;酶催化则以其高选择性、可进化性和环境友好性著称。将两者深度融合,有望突破天然酶催化的反应边界,实现全新的非天然生物催化转化。然而,目前的电酶催化策略大多局限于辅因子再生或级联反应,真正利用电场驱动酶的非天然自由基反应性仍充满挑战。
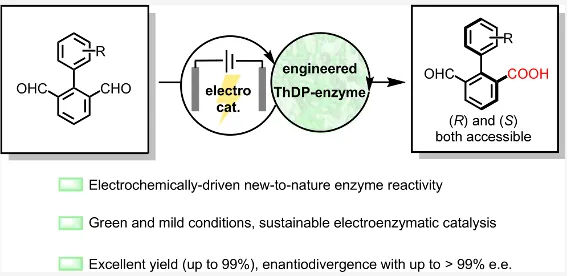
近期,南京大学黄小强课题组在电酶催化合成领域取得突破。他们将焦磷酸硫胺素(ThDP)依赖酶与阳极电氧化相结合,成功开发出电酶催化氧化去对称化新策略,实现了两类手性分子的高效不对称合成:轴手性联芳基化合物的对映选择性合成(J. Am. Chem. Soc.2026, DOI: 10.1021/jacs.6c01275)以及硅手性中心有机硅化合物的立体选择性构建(Angew. Chem. Int. Ed.2026, DOI: 10.1002/anie.3445735)。两项工作通过工程化改造ThDP依赖的苯甲醛裂解酶(PfBAL),结合易得的二茂铁甲醇(FcMeOH)作为电介质,在不分隔电解池中以恒定电流驱动反应。该方法展现出优异的底物普适性(联芳基底物17例,硅基底物22例)、极高的对映选择性(ee值高达>99.5%)、良好的产率(最高99%)和易于放大,并且酶用量可低至0.1 mol%,转化数(TON)最高达930。该体系避免了传统化学催化常见的过氧化与再对称化问题,为轴手性及硅手性分子的绿色合成提供了全新的生物催化平台。
轴手性联芳基化合物的对映选择性合成,JACS
手性中心有机硅烷的立体选择性构建,Angew
研究背景:从光酶到电酶
黄小强团队长期致力于开发新型酶催化元件和非天然生物合成体系。前期,他们先后实现了ThDP依赖酶的光驱动自由基酰化(Nature2024, 625, 74-78)、光酶催化对映选择性三自由基分选(Nature2025, 637, 1118-1123),并发展电驱动酶促动态动力学氧化(Nature2025, 643, 699-704),成功将ThDP依赖酶催化从天然的双电子极性翻转机制拓展至单电子自由基路径。
电酶催化氧化去对称化:轴手性联芳基的合成
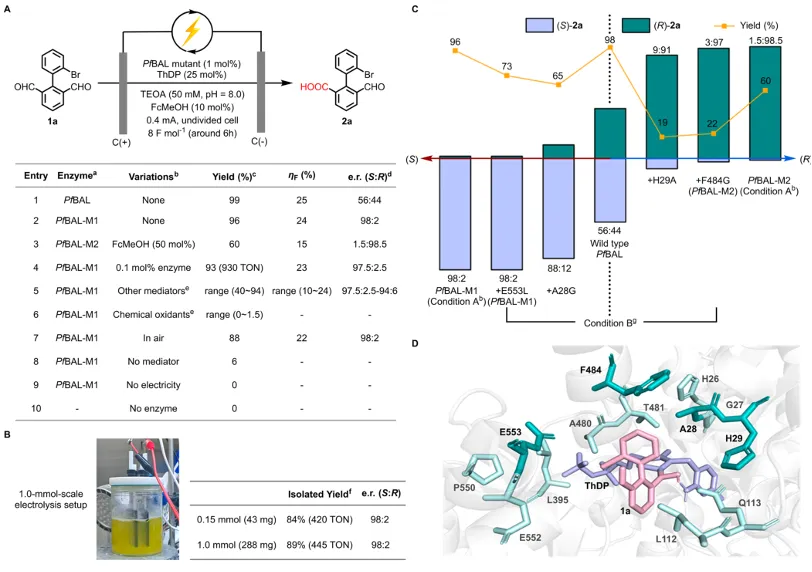
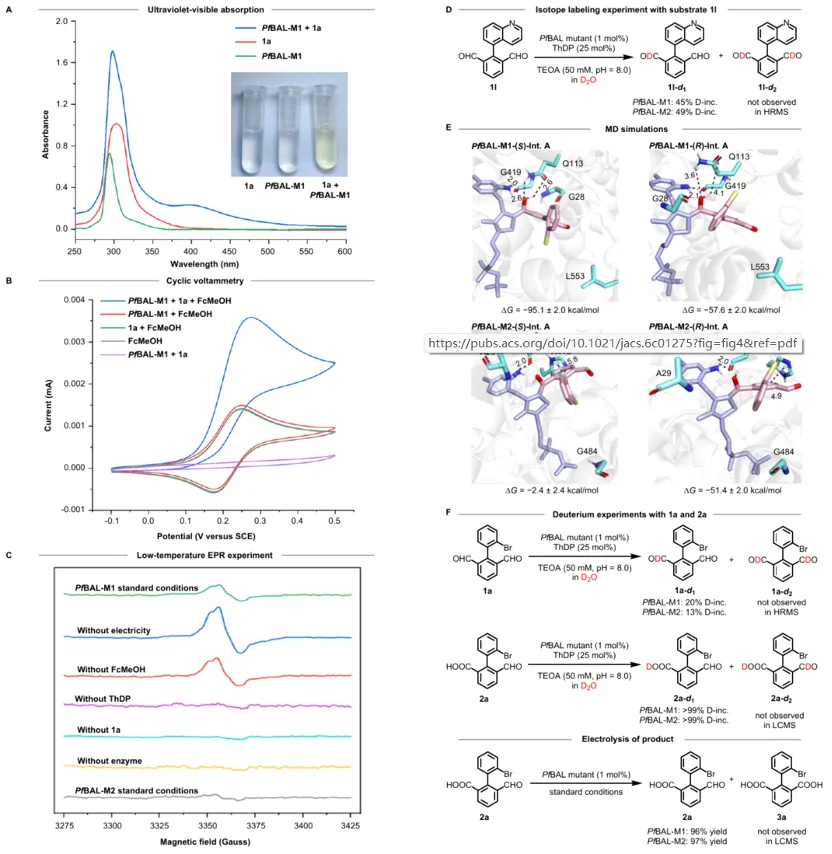
作者选择联芳基二醛(1a)作为模型底物,以PfBAL为起始酶,二茂铁甲醇(FcMeOH)为电化学介体,在石墨电极、恒定电流(0.4 mA)条件下进行反应。野生型PfBAL能以98%的产率和较差ee值得到产物。通过两轮定点突变,团队获得了两个高性能突变体:在弱碱性条件下(pH 8.0),PfBAL‑M1(A28G‑E553L)给出96%产率、ee值96%,同时PfBAL‑M2(H29A‑F484G)给出60%产率和97% ee。并且有趣的是,两个突变体分别给出相反构型的产物(S‑2a 和R‑2a),实现了对映发散性合成。控制实验表明,酶、电流、电介质三者缺一不可,且放大至1.0 mmol规模时,仅需0.2 mol%酶即可获得89%分离产率,ee值保持不变。
上图:轴手性联芳基合成的反应条件与酶突变体优化
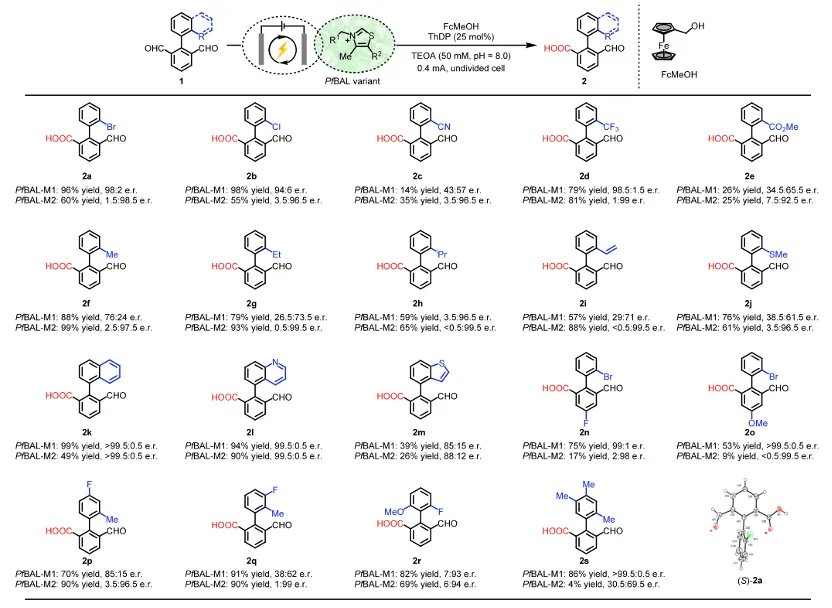
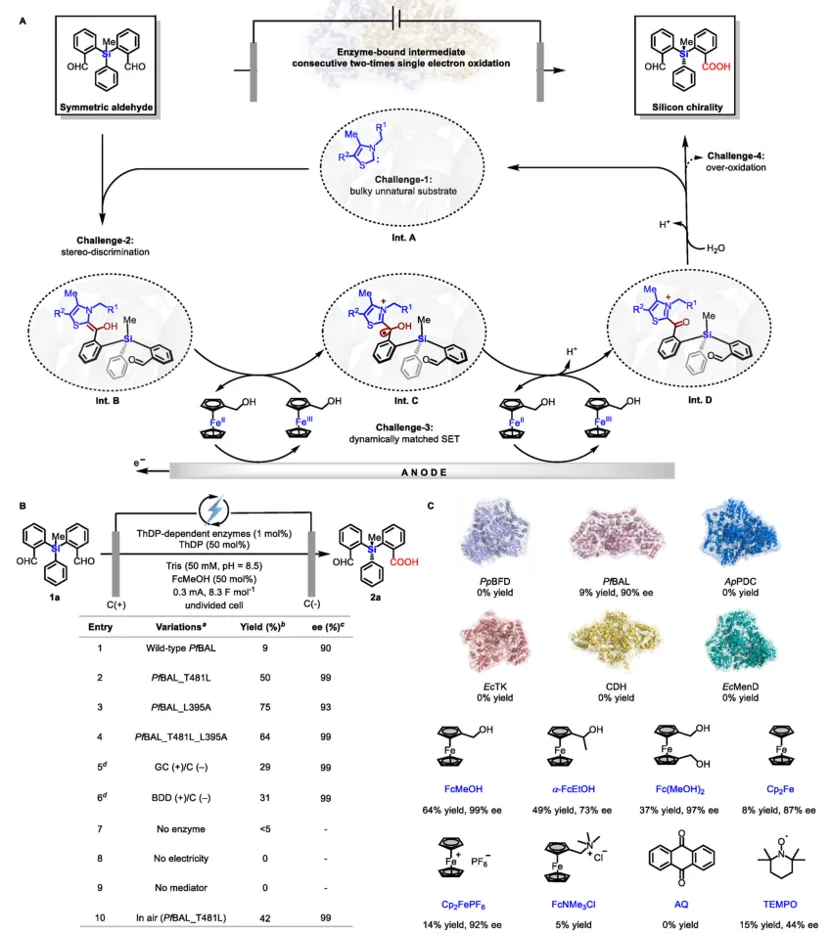
在底物范围方面,该电酶体系对多种对称联芳基二醛表现出优异的兼容性。苯环上无论是给电子(如甲氧基)还是吸电子(如三氟甲基)取代均能良好耐受。位阻较大的邻位甲基、萘环以及苯并杂环等底物同样反应顺利,ee值最高超过99%。二取代、三取代底物也能获得中等到良好的产率。与传统化学催化(如NHC催化)相比,本方法完全避免了二酸过氧化产物的生成,解决了该领域长期存在的痛点。
上图:电酶催化轴手性联芳基底物适用性
机理研究表明,底物二醛与ThDP辅因子在酶活性中心缩合形成Breslow中间体后,阳极氧化生成的FcMeOH⁺从中夺取一个电子,产生酰基自由基中间体(低温EPR实验捕获到了自由基信号),再经一次单电子氧化生成酰基噻唑阳离子,水解后释放手性羧酸产物并再生卡宾ThDP。分子动力学模拟显示,在PfBAL‑M1中,(S)-构型的Breslow中间体与活性口袋中的Q113、M421等残基形成更稳定的氢键网络,结合自由能更低;而在PfBAL‑M2中,(R)-构型则通过T型π-π堆积与H415相互作用获得优势。这一精细的非共价相互作用调控解释了两个突变体的对映发散性。此外,同位素标记实验表明,产物中的醛基氢无氘掺入,且将产物重新置于电酶条件下可定量回收,证明酶对产物醛基不再识别,从而有效阻断了二次氧化。
上图:电酶催化自由基反应机理与验证
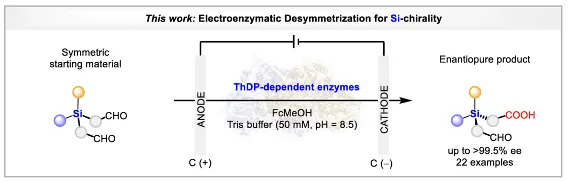
拓展至硅手性中心的构建
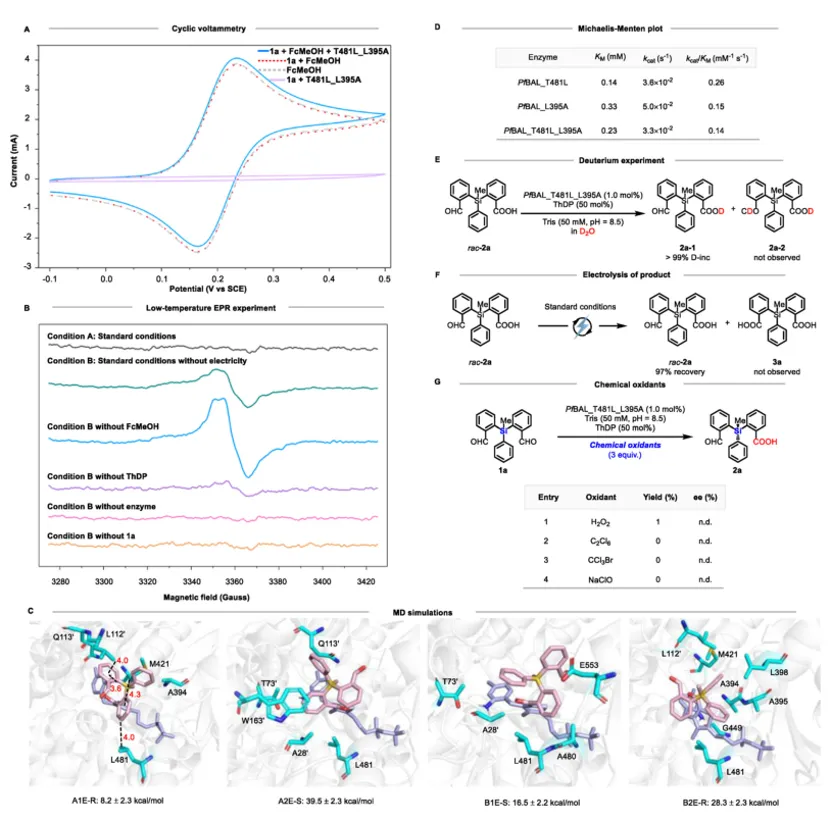
在上述轴手性工作基础上,团队进一步将电酶去对称化策略应用于硅手性中心的构建。以硅烷二醛(1a)为模型底物,野生型PfBAL仅给出9%产率和90% ee。通过定点突变,作者筛选出双突变体PfBAL_T481L_L395A,在优化条件下(pH 8.5,0.3 mA,50 mol% FcMeOH)获得64%产率、99% ee。控制实验再次确认酶、电流、电介质的必要性,且酶在0.3–1.0 mA电流范围内保持稳定,可承受高达8.3 F的电荷通量。
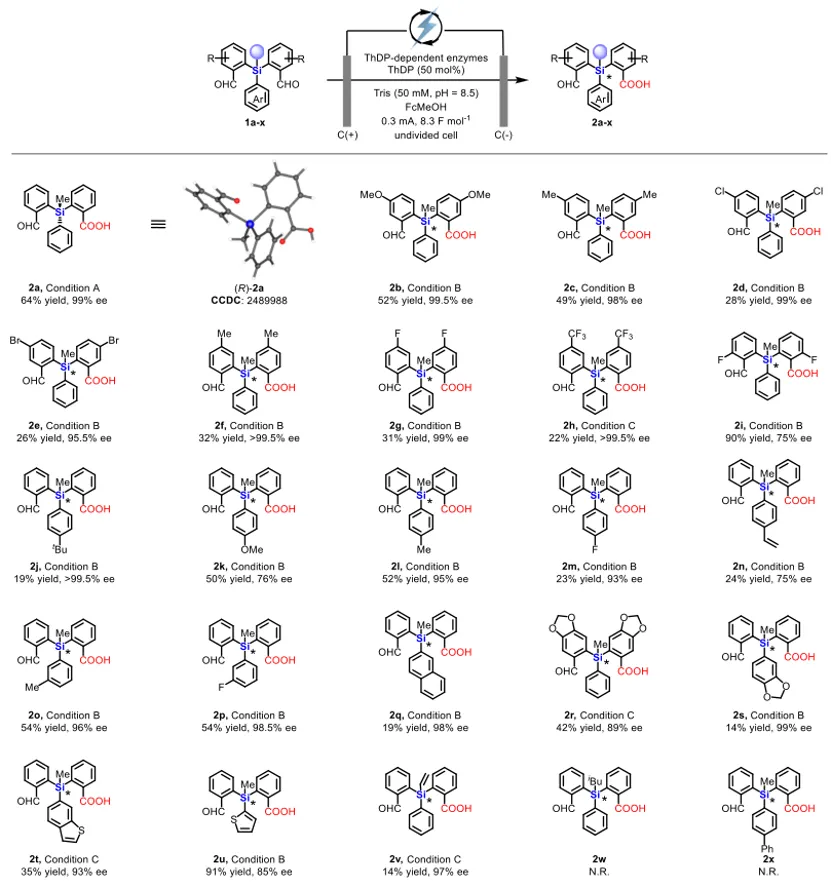
底物考察表明,22种对称硅烷二醛被成功转化,产物2a以(R)-构型为主(通过单晶衍射确认)。芳环上无论是给电子还是吸电子取代基均能兼容,氟、氯、溴等卤素原子不受影响,其中2f和2h的ee值达到>99.5%。位阻较大的叔丁基、萘基以及苯并噻吩等底物也可反应,虽然产率中等但ee值优异。烯基可以保留,氧化敏感基团不受影响。目前该方法对异丁基直接连于硅原子或苯环上有苯基取代的底物尚不兼容,这为后续酶工程化改造指明了方向。
上图:电酶催化硅手性中心底物范围
机理验证方面,循环伏安实验显示酶-底物-电介质三者共存时阳极峰电流升高、阴极峰降低,支持介导的单电子转移路径。低温EPR在无电解但有氧条件下检测到清晰的自由基信号,且信号依赖于酶、ThDP和醛底物,再次确认了酰基自由基中间体的存在。分子动力学模拟表明,AIE‑R构型的Breslow中间体与突变体活性口袋中的Q113’、M421、L112’等残基形成多重氢键和C=O···π、C–H···π相互作用,结合自由能最低,解释了(R)-选择性。产物2a在电酶条件下无任何氘掺入且可定量回收,再次证明酶对产物醛基无催化活性。
上图:电酶催化自由基反应机理与验证
黄小强团队通过将ThDP依赖酶与电化学介导单电子氧化深度融合,成功开发了两类新型电酶催化体系,实现了轴手性联芳基和硅手性中心的去对称化合成。该工作的核心创新在于:突破ThDP酶天然的双电子催化机制,利用电化学开发了非天然的自由基路径;通过定向进化和电化学参数的协同优化,实现对映选择性高达>99.5%;适用于联芳基和硅烷两类完全不同的底物家族,展示了平台的通用性;结合CV、EPR、MD模拟等手段,探索了电酶催化机理;可放大至毫摩尔级、酶载量低、无过氧化副反应,具备实际应用潜力。这两项工作将电化学合成从传统的辅因子再生工具升级为解锁酶非天然反应性的通用策略,为手性分子合成提供了一类生物催化解决方案。


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
