第一作者:Fanfan Lao
通讯作者:刘福强、朱长青
期刊:Applied Catalysis B: Environment and Energy
发表时间:2026 年 5 月 4 日 online
DOI:https://doi.org/10.1016/j.apcatb.2026.126907
近日,南京大学环境学院刘福强教授、生态环境部南京环境科学研究所朱长青副研究员等在 Applied Catalysis B: Environment and Energy 发表题为 “Single atom and adjacent cluster synergistically optimize non-radical thermodynamic pathway of peroxymonosulfate activation for efficient antibiotic wastewater detoxification” 的研究论文。
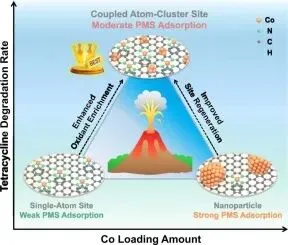
该工作围绕 PMS 高级氧化中一个非常关键的问题展开:单原子位点虽然原子利用率高,但 PMS 富集与活化能力有限;金属纳米颗粒虽然更容易吸附 PMS,却容易出现位点利用率低、吸附过强和中间体难脱附的问题。作者提出一种“单原子 CoN5 位点 + 邻近 Co4 团簇”的协同结构,在多金属团簇增强 PMS 捕获的同时,保留单原子位点高效反应与快速脱附的优势,从而实现四环素废水的高效、持久、低毒化处理。
单原子 CoN5 位点与邻近 Co4 团簇协同调控 PMS 吸附、活化和脱附过程,推动污染物通过表面碰撞氧化路径发生电子转移降解。
抗生素废水的难点,不只是“能不能降解”,更在于“能不能持续、低毒、面向真实水体地降解”。
以四环素为代表的抗生素在水环境中具有持久性和生态风险。PMS 基高级氧化过程可以提供强氧化能力,但在实际体系中,催化位点往往面临两个矛盾:如果 PMS 吸附太弱,表面氧化剂富集不足;如果吸附太强,反应后产物或 HSO4- 难以脱附,又会导致位点中毒。
这篇文章的核心思路,就是在单原子与团簇之间寻找一个平衡点。
作者以氮丰富的 MOF MIP-202 为前驱体,通过调控 Co 负载量和热解过程,构建了一系列 MCo-C-x 多孔碳催化剂。其中,MCo-C-12 形成了空间邻近的 CoN5-Co4 结构:Co 单原子位点保证高原子利用率和快速反应,邻近 Co4 团簇增强 PMS 捕获与表面氧化剂富集。二者协同后,体系不再依赖传统自由基路径,而是通过表面碰撞氧化路径实现污染物电子转移降解。
在性能上,MCo-C-12/PMS 体系对四环素表现出非常突出的降解效率,催化剂剂量归一化速率常数达到 17.70 L min-1 g-1,高于多数已报道体系 1-2 个数量级。更重要的是,固定床反应器中仅使用 100 mg 催化剂,即可在真实水体中连续实现四环素完全去除超过 312.5 h,显示出较强的应用潜力。
1. 用 MIP-202 构建单原子-团簇邻近位点,解决 PMS 活化中的吸附-脱附矛盾
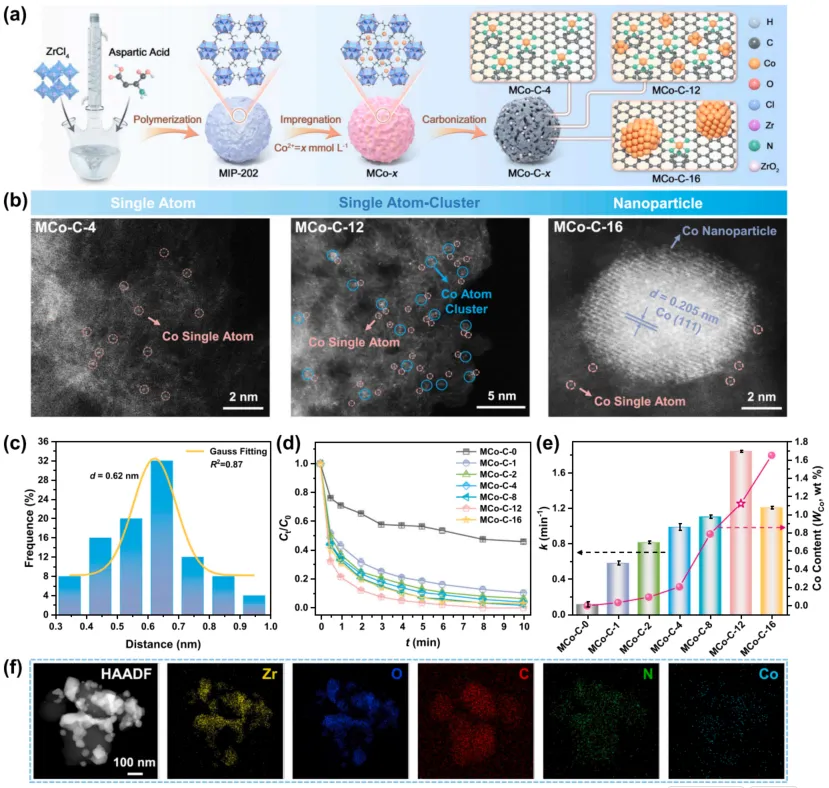
作者首先以 MIP-202 为前驱体制备 MCo-C-x 催化剂。MIP-202 的小孔径和含氮配体有利于稳定 Co 原子;当 Co 负载量较低时,主要形成孤立 Co 单原子;当 Co 负载量增加时,部分 Co 原子在孔道限域作用下形成亚纳米团簇,并与单原子位点保持邻近。
在这一系列样品中,MCo-C-12 表现最优。HAADF-STEM 结果显示,其同时具有 Co 单原子和亚纳米 Co 团簇,且两类位点在空间上相互接近。相比纯单原子位点或纳米颗粒位点,CoN5-Co4 的优势在于同时兼顾 PMS 富集、活化和反应后脱附。
图 1. MCo-C-x 催化剂的合成示意、HAADF-STEM 表征、Co 单原子与 Co 团簇距离统计、四环素降解性能及元素分布。结果表明,调控 Co 负载量可以获得单原子、单原子-团簇邻近结构和纳米颗粒等不同 Co 物种,其中 MCo-C-12 的 CoN5-Co4 邻近位点带来最优 PMS 活化性能。
2. CoN5-Co4 位点显著提升四环素降解速率,并在真实水体连续运行中保持稳定
在 PMS 活化降解四环素实验中,MCo-C-12 表现出最快的降解效率。不同 Co 负载样品在 10 min 内的四环素去除率随 Co 结构演化而明显变化,其中 MCo-C-12 可实现 100% 去除,催化剂剂量归一化速率常数达到 17.70 L min-1 g-1。
这一数值的意义在于,它不是简单比较表观速率,而是将催化剂投加量纳入考虑后得到的活性指标。也就是说,MCo-C-12 不是靠“多加催化剂”实现高去除,而是位点本身具有更高的 PMS 活化效率。
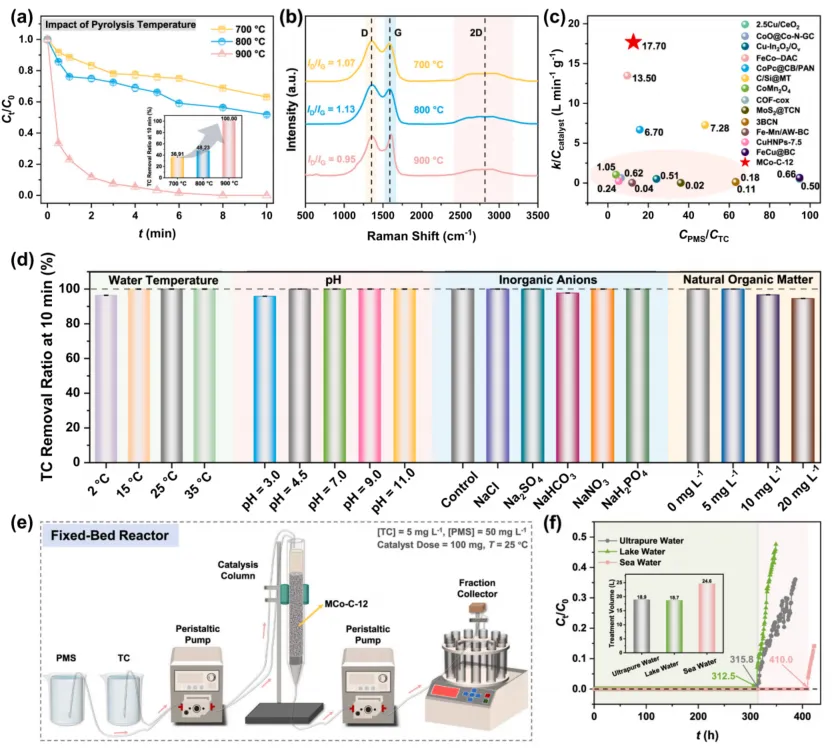
作者还进一步考察了温度、pH、无机阴离子、天然有机质和真实水体条件。MCo-C-12/PMS 在 2-35 摄氏度和 pH 3.7-9.2 范围内均能保持较好性能。固定床反应器实验中,100 mg 催化剂在真实水体中实现四环素连续完全去除超过 312.5 h,说明该体系不只停留在烧杯实验层面。
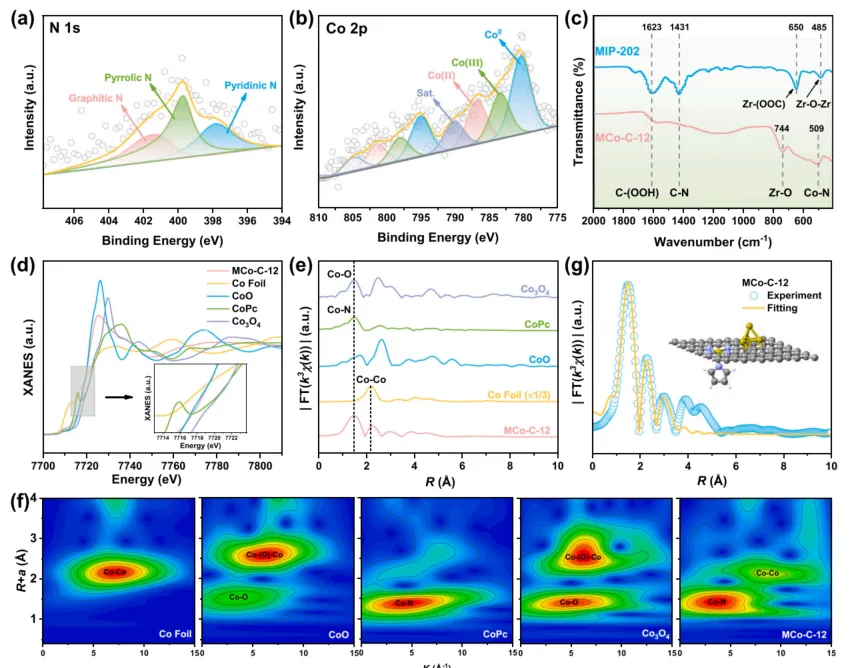
图2. MCo-C-12 的 N 1s、Co 2p XPS,FT-IR,Co K-edge XANES/EXAFS,小波变换及拟合结果。表征结果证明,MCo-C-12 中 Co 主要以 CoN5 单原子和邻近 Co4 团簇形式存在,为后续 PMS 吸附、活化和脱附协同提供结构基础。
图 3. 不同热解温度下催化剂的四环素降解性能、Raman 结构分析、与文献催化剂的活性对比、环境因素影响以及固定床连续运行结果。MCo-C-12/PMS 在复杂水质和长时间连续流条件下仍保持高效四环素去除能力。
3. 主导路径不是传统自由基,而是表面碰撞氧化引发的电子转移
高级氧化研究中,一个常见问题是:体系到底靠自由基,还是靠非自由基?
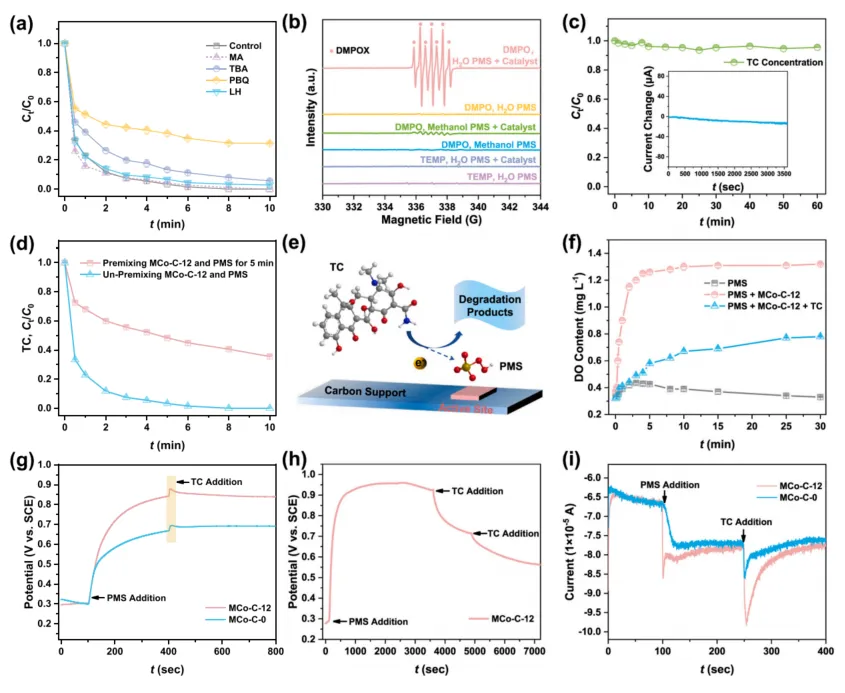
这篇文章通过淬灭实验、ESR、电化学测试和预混实验对反应路径进行了系统判断。结果显示,常见自由基捕获剂对四环素降解影响有限,说明 SO4·- 和 ·OH 并不是主要贡献者。TEMP 和 DMPO 相关 ESR 测试表明体系中存在 1O2 信号,但文章进一步指出,单靠 1O2 仍不足以解释整个反应行为。
更关键的是,预混 MCo-C-12 和 PMS 后再加入四环素,降解效率出现明显变化;OCP 与 i-t 曲线也显示 MCo-C-12/PMS 体系具有更强的界面电子转移能力。作者据此提出,PMS 首先在 CoN5-Co4 位点上形成高电势表面复合物,当四环素与该复合物发生表面碰撞时,电子由污染物转移至表面氧化剂,从而实现非自由基氧化。
这一路径被作者称为 surface collision oxidation path,即表面碰撞氧化路径。
图 4. 淬灭实验、ESR 谱图、预混实验、表面碰撞氧化路径示意、溶解氧变化以及电化学测试。结果表明,MCo-C-12/PMS 体系主要通过表面结合 PMS 复合物诱导污染物电子转移,而非依赖传统自由基链式氧化。
4. 降解不是终点,低毒化和可生物利用性同样重要
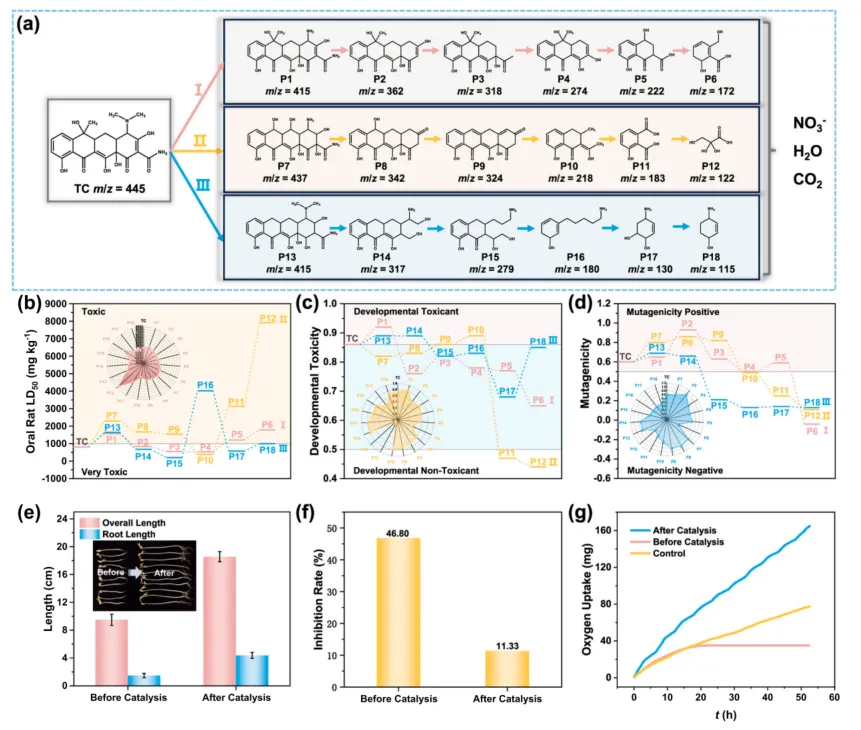
抗生素降解体系经常会遇到一个现实问题:母体污染物降低了,但中间产物毒性未必同步下降。作者因此对四环素降解路径、产物毒性和微生物响应进行了进一步评估。
LC-MS 推断显示,四环素在 MCo-C-12/PMS 体系中经历脱甲基、羟基化、开环、脱羧和进一步矿化等过程。毒性预测结果表明,反应后产物的急性毒性、发育毒性和致突变风险整体下降。
实验层面的证据也支持这一结论。发光菌测试中,原始四环素溶液对发光菌的抑制率为 46.80%,经催化处理后下降至 11.33%。污泥呼吸实验进一步说明,处理后的产物更容易被微生物利用,具有从“降解”走向“解毒”和“可后续生化处理”的意义。
图 5. 四环素可能降解路径、产物毒性预测、植物萌发测试、发光菌毒性测试和污泥呼吸实验。结果表明,MCo-C-12/PMS 不仅能够高效去除四环素,还能降低反应产物生态毒性,并提升后续微生物利用可能性。
5. DFT 揭示协同本质:团簇帮助 PMS 富集,单原子保证位点再生
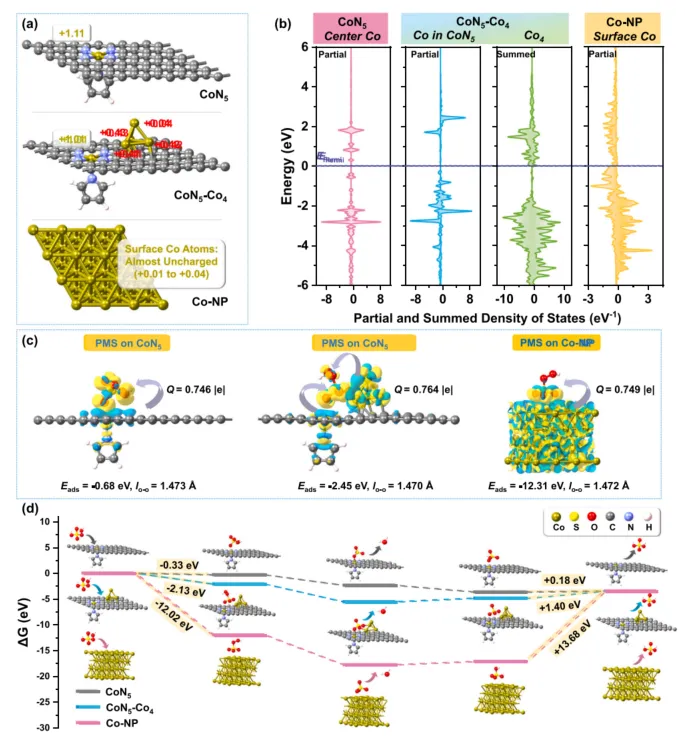
为了理解 CoN5-Co4 为什么优于单原子 CoN5 和 Co 纳米颗粒,作者进一步进行了理论计算。
结果显示,单原子 CoN5 对 PMS 的吸附相对较弱,不利于表面 PMS 富集;Co 纳米颗粒对 PMS 及中间体吸附过强,容易造成位点占据和反应迟滞。相比之下,CoN5-Co4 结构处在更合适的区间:邻近 Co4 团簇增强 PMS 捕获和电子调控,CoN5 单原子位点则有利于反应后 HSO4- 快速脱附和活性位点再生。
换句话说,MCo-C-12 的高活性并不是单一结构贡献,而是“吸附得上来、反应得出去、位点能恢复”的整体平衡。
图 6. CoN5、CoN5-Co4 和 Co 纳米颗粒模型的 Bader 电荷、DOS、静电势、PMS 吸附构型、Gibbs 自由能及结构演化。计算结果说明,CoN5-Co4 位点能够在 PMS 富集、表面氧化剂形成和 HSO4- 脱附之间取得更优热力学平衡。

- 提出单原子-邻近团簇协同活化 PMS 的结构策略。 文章不是简单比较单原子和纳米颗粒,而是构建 CoN5-Co4 邻近结构,用团簇增强 PMS 捕获,用单原子保证高效反应和位点再生。
- 将 PMS 活化性能提升到高水平。 MCo-C-12 对四环素降解的催化剂剂量归一化速率常数达到 17.70 L min-1 g-1,高于多数已报道催化剂 1-2 个数量级。
- 明确非自由基表面碰撞氧化路径。 体系主要通过表面结合 PMS 高电势复合物与污染物碰撞后发生电子转移,而非依赖传统 SO4.- 或 .OH 自由基主导氧化。
- 关注抗生素废水的“解毒”而不仅是“去除”。 四环素经处理后毒性显著降低,发光菌抑制率由 46.80% 降至 11.33%,并显示出更好的微生物可利用性。
- 具备连续流真实水体验证。 固定床反应器中,100 mg 催化剂可在真实水体中实现四环素连续完全去除超过 312.5 h,增强了该体系面向实际应用的说服力。

这篇 ACB 的价值,在于把 PMS 活化中的“位点设计”讲得更细。
过去我们常说单原子催化剂原子利用率高、纳米颗粒吸附能力强,但二者各自都有短板。单原子可能吸附不够强,颗粒又可能吸附过强、脱附困难。本文提出的 CoN5-Co4 邻近结构,正好把两者的优势放在同一个反应单元中:团簇负责把 PMS 抓住并调控电子结构,单原子负责高效反应和活性位点再生。
这种设计思路对高级氧化催化剂开发很有启发。真正高效的 PMS-AOPs 位点,不只是“更强氧化”或“更多活性中心”,而是要在吸附、活化、电子转移、产物脱附和长期稳定之间找到一个热力学与动力学都合适的平衡点。
https://doi.org/10.1016/j.apcatb.2026.126907
本文仅用于推广高级氧化前沿技术,如有侵权,请联系删除。