—— literature interpretation ——文献解读
标题:全基因组交叉性状分析描述了肺部疾病与胃肠道疾病之间共享的遗传结构
发表期刊:Nature Communications
发表时间:2025年3月
影响因子:15.7/Q1
肠道-肺轴介导脏器间炎症交互,遗传多效位点是呼吸与消化共病重要诱因。然而调控两类疾病共病的关键易感基因与菌群介导通路尚不明确。
人群数据:整合欧、亚、非多族群GWAS公共汇总数据开展肺-胃肠疾病遗传分析。
生物信息学:LDSC、MTAG等算法筛选多效SNP与候选易感基因。
检测技术:TWAS/PWAS联合孟德尔随机化、肠道菌群中介分析解析致病通路。
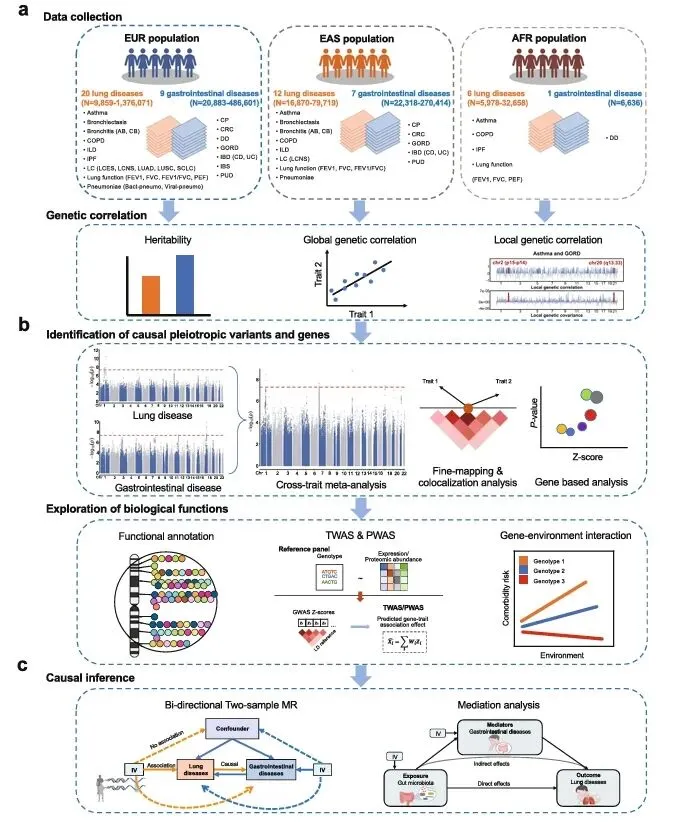
研究整合欧、东亚、非洲三大人群多系统GWAS数据,分三阶段分析:LDSC评估全基因组及局部遗传相关;跨性状Meta分析、精细定位与共定位筛选多效变异,联合TWAS/PWAS挖掘候选基因;双向MR与中介分析引入肠道菌群,系统解析肺-胃肠共病的共享遗传基础与肠肺轴因果通路(图1)。
复现建议:收集欧/东亚/非洲人群肺病、胃肠病GWAS汇总数据。依次执行LDSC遗传相关、MTAG荟萃、精细共定位、MAGMA基因富集、TWAS/PWAS及两样本MR菌群中介分析。使用LDSC、coloc、TwoSampleMR等软件。多重检验用Bonferroni校正。
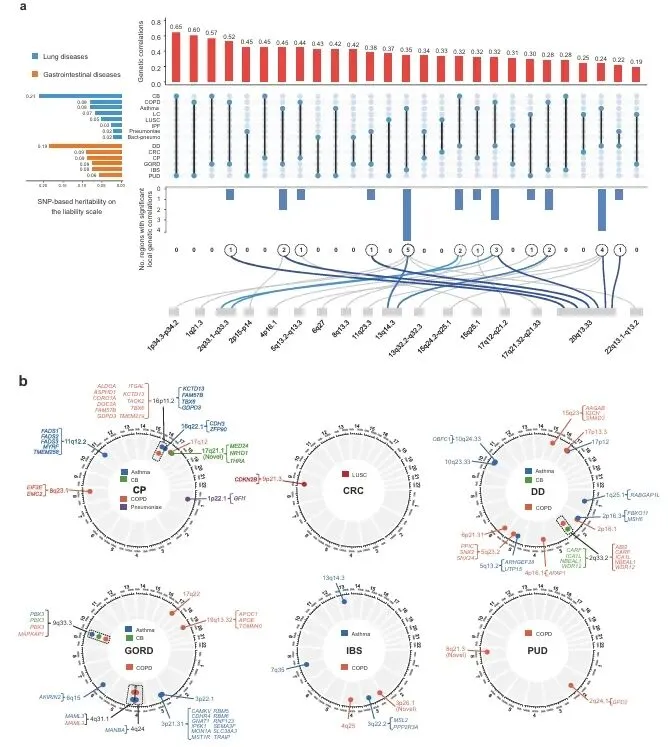
欧洲人群经校正后鉴定27组显著遗传相关配对,慢性支气管炎与消化性溃疡相关性最高(rg=0.65)。全基因组定位24个局部共享区段,2q33.2等区域在多病重叠。精细定位获42个候选SNP,其中rs55673000等3个为新变异,环状图展示染色体位点分布规律(图2)。
复现建议:LDSC计算全基因组rg,SUPERGNOVA做区段关联。MTAG跨性状荟萃后精细定位,coloc共定位筛选候选SNP(PPH4≥0.5)。GPA/PLACO交叉验证。R绘制Circos及UpSet图。统计用Bonferroni校正。
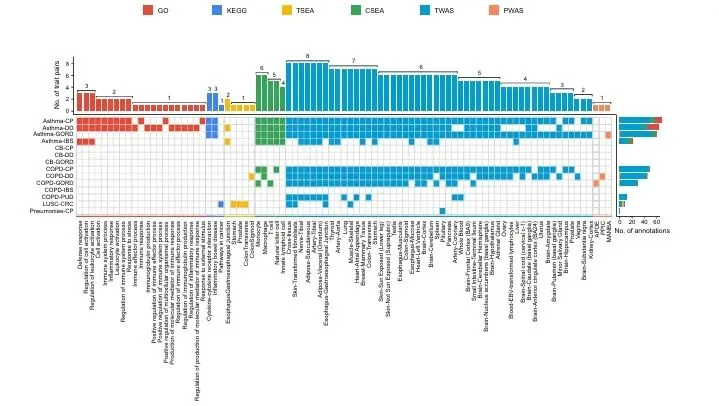
42个候选变异锚定66个共享多效基因。GO/KEGG富集于免疫炎症应答;组织富集显示基因集中于胃、结肠等消化道;细胞富集以T淋巴细胞为主。TWAS证实58个基因表达变化与共病风险显著相关,PWAS筛选出血浆MANBA、PPIC、APOE蛋白与发病显著关联(图3)。
复现建议:基于候选基因列表,clusterProfiler完成GO/KEGG富集。deTS、WebCSEA分别做组织、细胞富集。FUS依托GTEx/ARIC开展TWAS/PWAS分析。FDR<0.05为阈值。
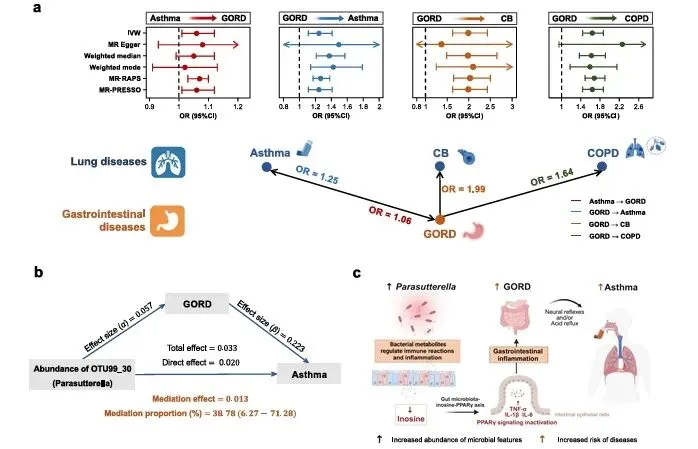
双向MR明确4组正向因果,哮喘↔胃食管反流为双向因果,胃食管反流分别诱发慢性支气管炎与慢阻肺。中介分析证实Parasutter菌介导胃食管反流升高哮喘风险,Faecalibacterium介导胃食管反流至慢支。机制上Parasutter下调肌苷、抑制PPARγ、升高促炎因子,完成级联致病(图4)。
复现建议:筛选显著IV工具变量,TwoSampleMR包行IVW/Egger/加权中位数双向MR。MR-PRESSO剔除异常SNP。基于宿主-菌群GWAS做两步MR+中介分析,delta法计算中介效应与占比。FDR校正P值。
本研究通过跨性状全基因组分析筛选出66个肺肠共病多效基因,多富集免疫炎症通路,并证实Parasutterella菌经胃食管反流介导哮喘发病,为肺肠轴共病遗传机制与防控提供遗传标志物。





 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
