第一作者:Shi Wang、Xinzhu Jiang
通讯作者:河海大学 杨汉培、南京理工大学张根
论文DOI:https://doi.org/10.1002/adma.73727
光催化双电子氧还原反应(2e− ORR)为绿色合成过氧化氢(H₂O₂)提供了一条可持续的途径。然而,其效率在本质上受到质子转移与异质界面间电子迁移之间动力学不匹配的限制。受天然氢化酶中质子-电子协同迁移的启发,报道了一种邻苯二酚-三嗪供体-受体(D-A)共价有机框架材料——2,3-Dhta-Tt,用于光催化制备H₂O₂时实现定向协同质子-电子转移(DCPET)。该材料固有的内建电场结合邻苯二酚衍生的动态质子接力网络,可对质子和电子流进行定向排列,并建立周期性共输运通道,将质子输送至三嗪受体位点。在分子层面,儿茶酚供体单元占据最高占空轨道(HOMO),同时作为光激发中心和初始质子释放位点,从而实现质子传递与电子迁移的同步。这种矢量耦合降低了O—O氢化反应的活化能垒,促进了高选择性的2e− ORR,使纯水中H₂O₂的生成速率达到了27.22 mmol g⁻¹ h⁻¹。该框架还表现出6.09 × 10⁻⁻⁵ S cm⁻¹的质子电导率和94.45 ps的延长激发态寿命。同位素标记、原位光谱学以及密度泛函理论(DFT)计算支持了质子循环过程以及质子/电子的定向参与。这项工作推动了异相光催化剂的设计,超越了传统的PCET协同效应。
图 1.设计逻辑 + 电子结构/静电势/孔道环境
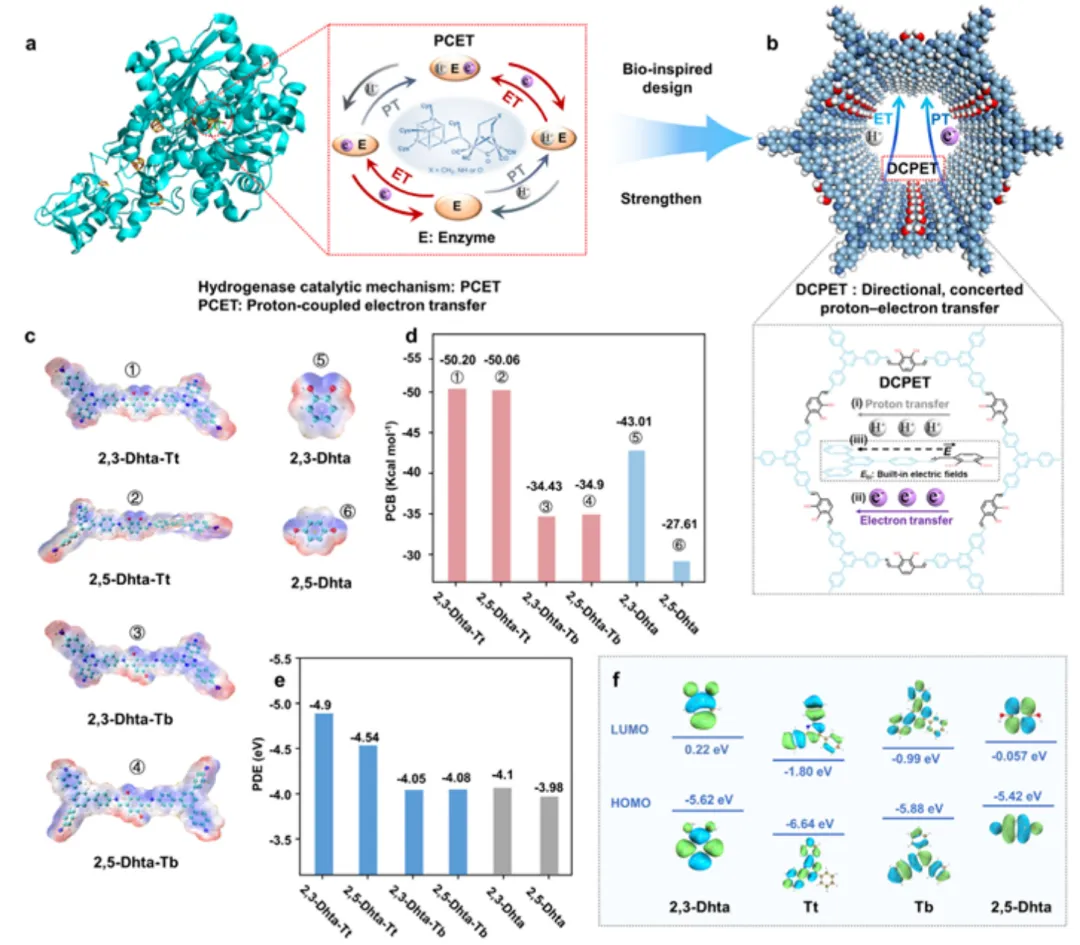
(a) 天然氢化酶 PCET 机器示意(质子中继/电子传递/活性位点空间组织);(b) COF 中的仿生 DCPET 概念模型:catechol 给体(donor)→ triazine 受体(acceptor)构成 D-A 通道,邻位二羟基通过氢键网络形成质子中继,内建电场提供方向性偏置。(c) ESP(静电势)映射:单体与 COF 中最负的 ESP 区域集中在邻位二羟基 O 附近,说明电子密度富集、O–H 键极化增强,有利于质子解离;(d) BCP(键临界点)分析可视化邻位 O–H···O 氢键处的电子密度堆积;(e) 羟基衍生质子中继位点的 PDB 能量随 ESP 单调下降,量化邻苯二酚排列对"质子释放倾向"的电子结构优势。(f) 前线轨道(HOMO/LUMO):HOMO 主要落在 catechol 给体 π 体系,LUMO 位于 triazine 中心区,给出 donor→acceptor 的轨道级预组织。
图 2. 光催化产 H₂O₂ 性能与表征集证
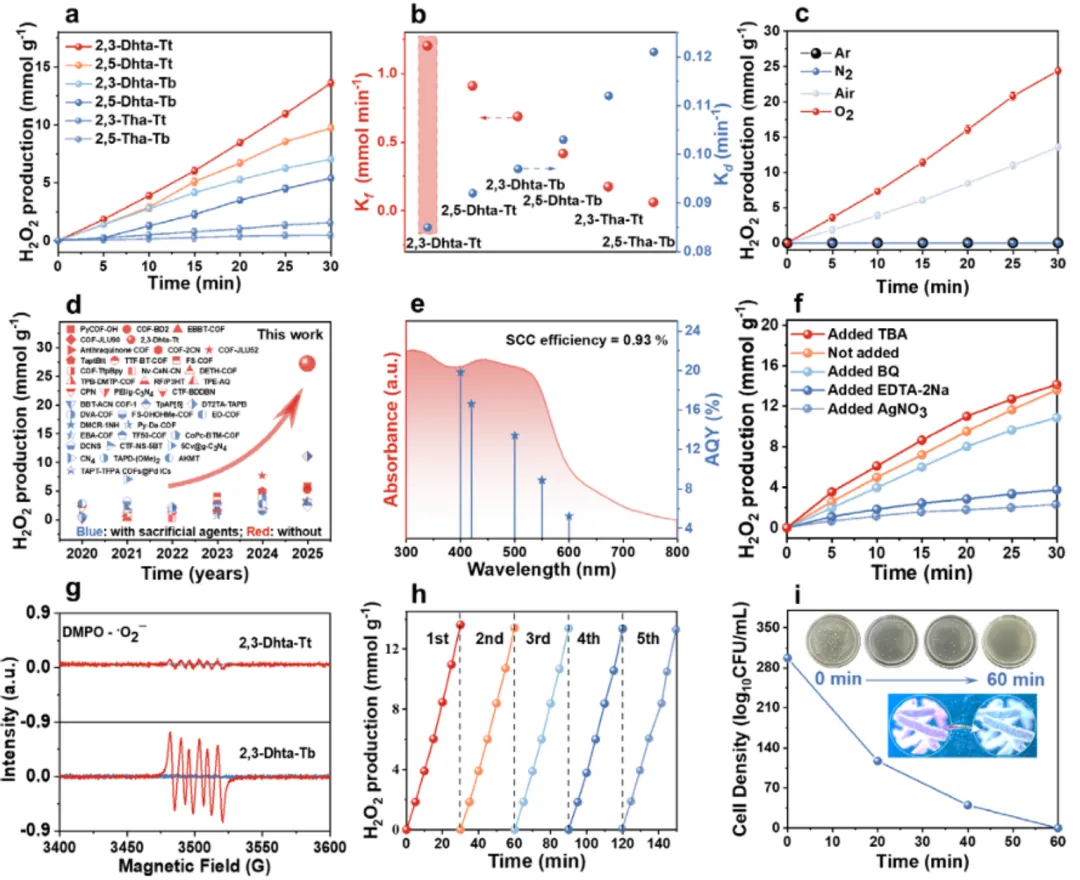
(a) 不同 COF 在空气饱和纯水中的 H₂O₂ 产率对比(2,3-Dhta-Tt 达 27.22 mmol·g⁻¹·h⁻¹,近十倍于对照 2,3-Tha-Tt);(b) 表观生成/分解速率常数(Ky / Kd)关系,说明 2,3-Dhta-Tt 能在"生成-分解"竞争中维持净积累;(c) 气氛对照(O₂ / N₂ / Ar)证明反应依赖分子 O₂;(d) 与文献基准体系的 H₂O₂ 产率/体系条件对比汇总(突出其在无牺牲剂纯水体系中的位置);(e) AQY@400 nm = 19.82%,SCC = 0.93%;(f) 电子清除(AgNO₃ / Na₂S₂O₈)+ 空穴清除(EDTA-2Na)实验表明光生 e⁻/h⁺ 均参与;(g) EPR 自旋捕获:仅有弱 ·O₂⁻ 信号,不支持自由基主导路径;(h) 循环稳定性;(i) 原位抗菌/降解应用示意(E. coli 灭活、抗生素/染料降解)。
图 3. D-A 电子结构、能带/表面势与载流子动力学
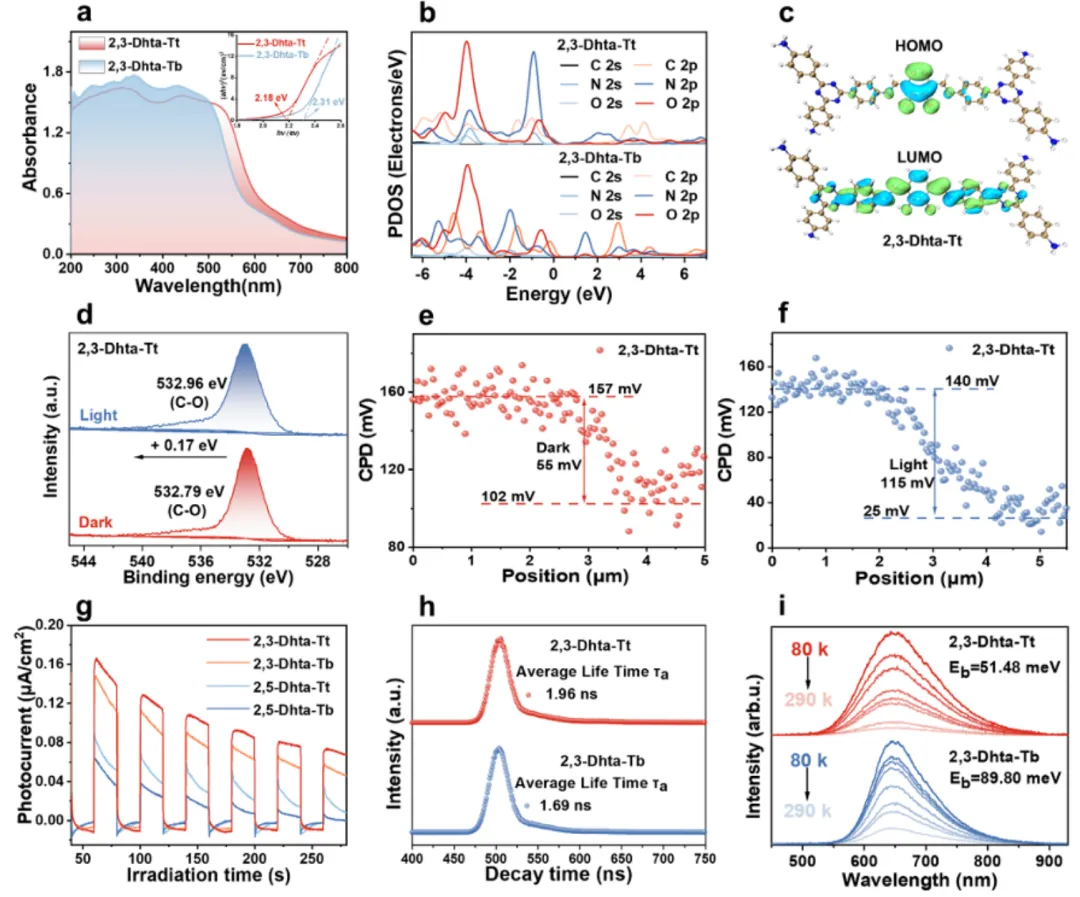
(a) UV–vis DRS 吸收边:2,3-Dhta-Tt 相对 2,3-Dhta-Tb 红移,Tauc 带隙 Eg ≈ 2.18 eV;(b) PDOS:价带顶主要来自 O 2p / C 2p(catechol),导带底主要来自 N 2p / C 2p(triazine),给出电子-质子功能区的轨道归属;(c) 实空间 HOMO/LUMO 分布(HOMO 在 catechol,LUMO 在 triazine),体现 D-A 预组织;(d) XPS O 1s 在光照下的偏移,反映邻位 –OH 参与光诱导电子激发;(e–f) KPFM 表面势差:2,3-Dhta-Tt Δ≈55 mV(光照后进一步放大),显著高于 2,3-Dhta-Tb(≈22 mV),定量其更强的光致内建电场与方向性电荷分离驱动力;(g) 瞬态光电流(最高 jph)与 (h) TRPL/寿命、(i) 温度依赖 PL 给出的激子束缚能 Eb(2,3-Dhta-Tt ≈51.5 meV vs Tb ≈89.8 meV),共同证明 D-A 极化促进激子解离与载流子寿命延长。
图 4. fs-TA 激发态动力学 + 同位素/质子传导/operando DRIFTS
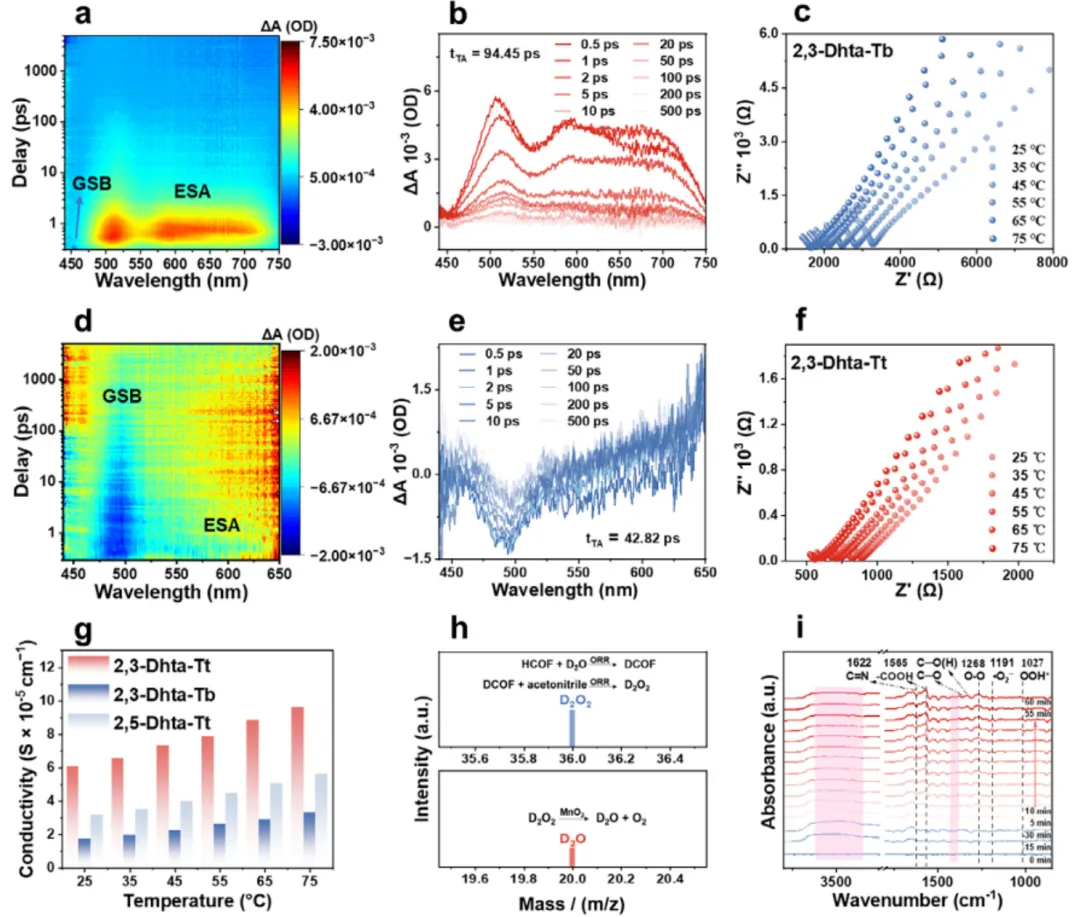
(a–b) 2,3-Dhta-Tt 的 fs-TA 谱:500–700 nm 宽正 ESA 带(光致分子内电荷转移/电荷分离态:空穴在 catechol、电子在 triazine),无明显负 GSB(因谱重叠+高效 CS depopulation);(c) 阻抗谱得出的质子电导 σ ≈ 6.09×10⁻⁵ S·cm⁻¹(25 °C,≈98% RH),Arrhenius 给出低 Ea 符合 Grotthuss 质子跳跃机制;(d–f) 对照 COF 的 TA/电导对比;(g) 质子电导/活化能汇总;(h) 同位素标记-MS:在 CH₃CN 中用 D-COF/D₂O 体系检测到产物侧 m/z = 36(D₂O₂) 与分解后 m/z = 20(D₂O),证实 catechol 单元可作为局域质子储库参与/交换质子进入 H₂O₂ 形成;(i) in situ/operando DRIFTS:光照下 1315 cm⁻¹(C–O(H₂)⁺ 质子化)/1292 cm⁻¹(C–O neutral)动态增强(catechol 质子化-去质子化循环),3471 cm⁻¹ O–H 消耗(水参与补质子),1268/1565 cm⁻¹(OOH / 表面 C=O)增长与 1191 cm⁻¹ 弱 ·O₂⁻ 信号,整体支持以直接 2e⁻ ORR/ OOH 路径为主、catechol 质子中继闭环循环的机制。
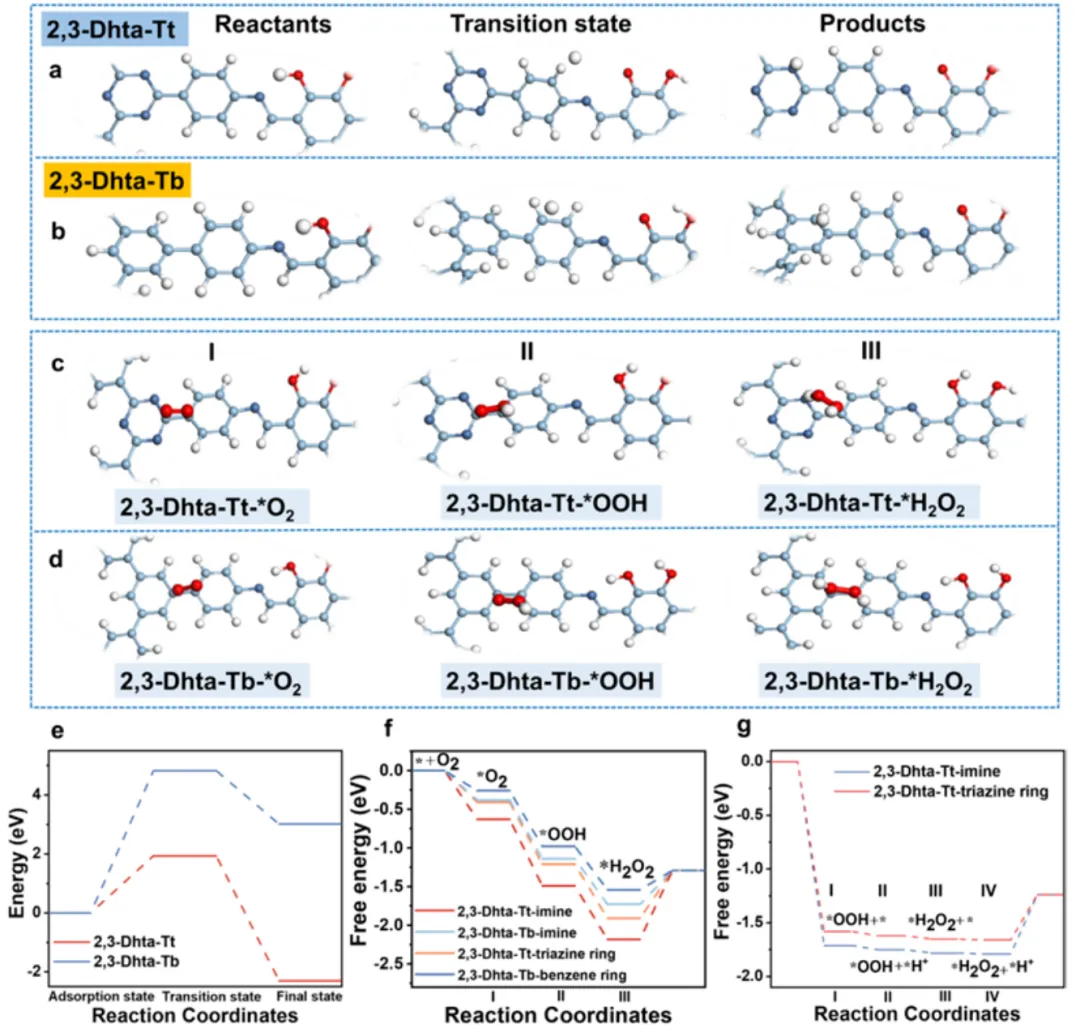
图 5. DFT 机理:质子迁移势垒 + ORR 自由能面 + DCPET 循环卡通
(a–b, e) 过渡态计算给出的质子迁移路径与能垒:在 2,3-Dhta-Tt 内建电场辅助下,质子从 catechol 源沿氢键/极性通道定向输送到 triazine/imine 活性位点,其关键质子活化能显著降低(triazine N 位点约 1.94 eV),远优于缺三嗪 D-A 的 2,3-Dhta-Tb(≈4.62 eV)。(c–d, f) ORR→H₂O₂ 的 DFT 反应自由能剖面:2,3-Dhta-Tt 的 OOH 形成 ΔG 更有利(文中给出约 −1.71 eV 级别的描述),后续步为放热级联;imine 与 triazine 相邻位点通过 D-A 链路协同稳定 OOH。(g) DCPET 循环概图:光激发 ⇒ e⁻ 沿 D→A 到 triazine;邻苯二酚 –OH ⇌ –O⁻ 释放质子沿中继到达同一 O₂ 吸附位点 ⇒ 同步质子-电子参与生成 OOH → H₂O₂;catechol 再质子化(补水/WOR 循环)完成闭合质子循环。
Directional Concerted Proton-Electron Transfer in COFs for Efficient Photocatalytic H2O2 Production
https://doi.org/10.1002/adma.73727

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
