肿瘤细胞“策反”免疫细胞来帮自己转移?ENO2是这个“策反计划”的总指挥
今天分享一项近期发表在《Signal Transduction and Targeted Therapy》上的重磅研究。南京医科大学第一附属医院孙跃明团队联合多家机构,通过对配对的原发灶、癌旁组织和肝转移灶进行单细胞RNA测序,结合空间转录组、基因编辑小鼠模型、患者来源类器官和虚拟筛选等技术,首次揭示:ENO2高表达的结直肠癌细胞,通过直接结合并稳定MIF蛋白,策反巨噬细胞向促癌的M2型极化,从而“借刀杀人”促进肝转移。而一种名为吡硫醇(pyrithioxin)的小分子抑制剂,能有效拆散ENO2-MIF复合物,在小鼠模型中显著抑制肝转移。
单细胞测序、空间转录组生信发文有诀窍!关注生信速通后台咨询,轻松拿下高分医学论文!
已整理好全套26年生信发文套路,后台可领取!
👇关注生信速通,带你拆解高分期刊
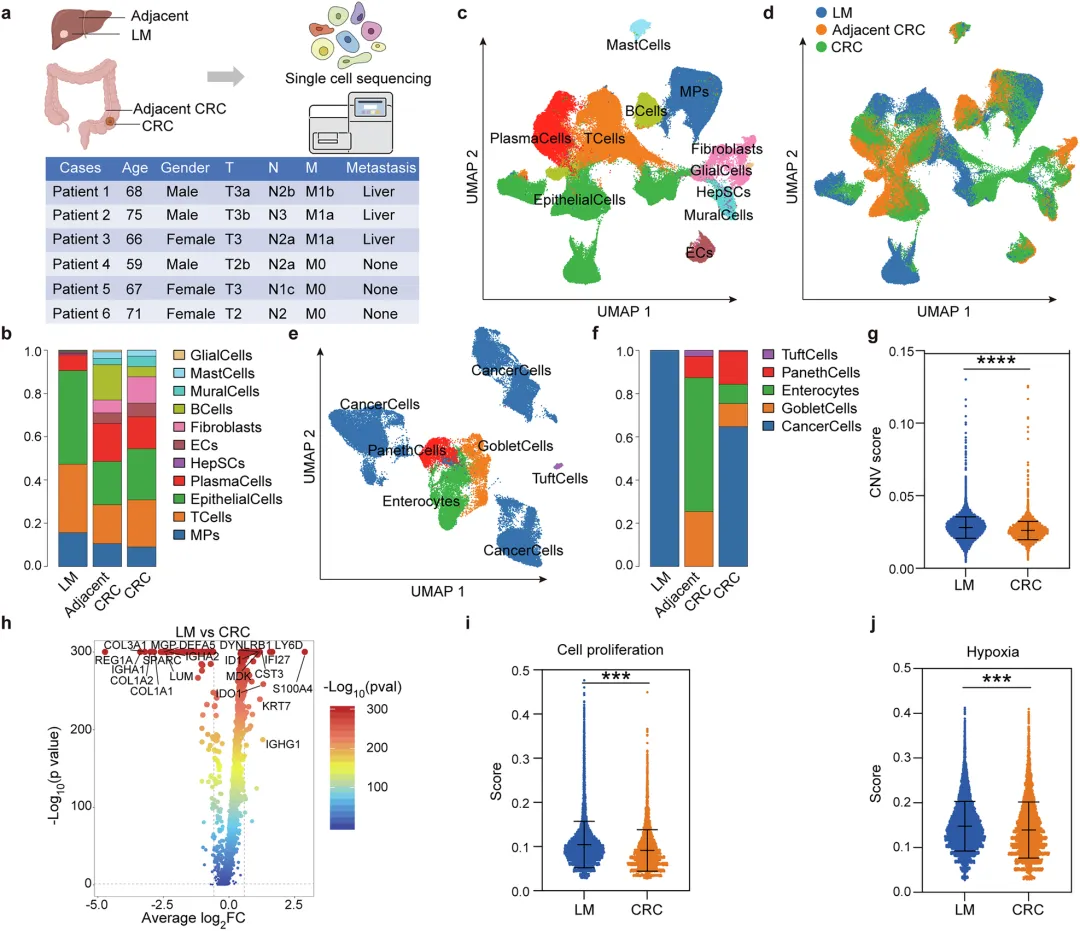
📌单细胞图谱锁定“转移特供”亚群:对3例CRLM患者和3例非转移CRC患者的15份配对样本进行scRNA-seq,发现ENO2⁺癌细胞在转移患者中显著富集,且高表达EMT相关基因,是“转移启动状态”的核心标志。
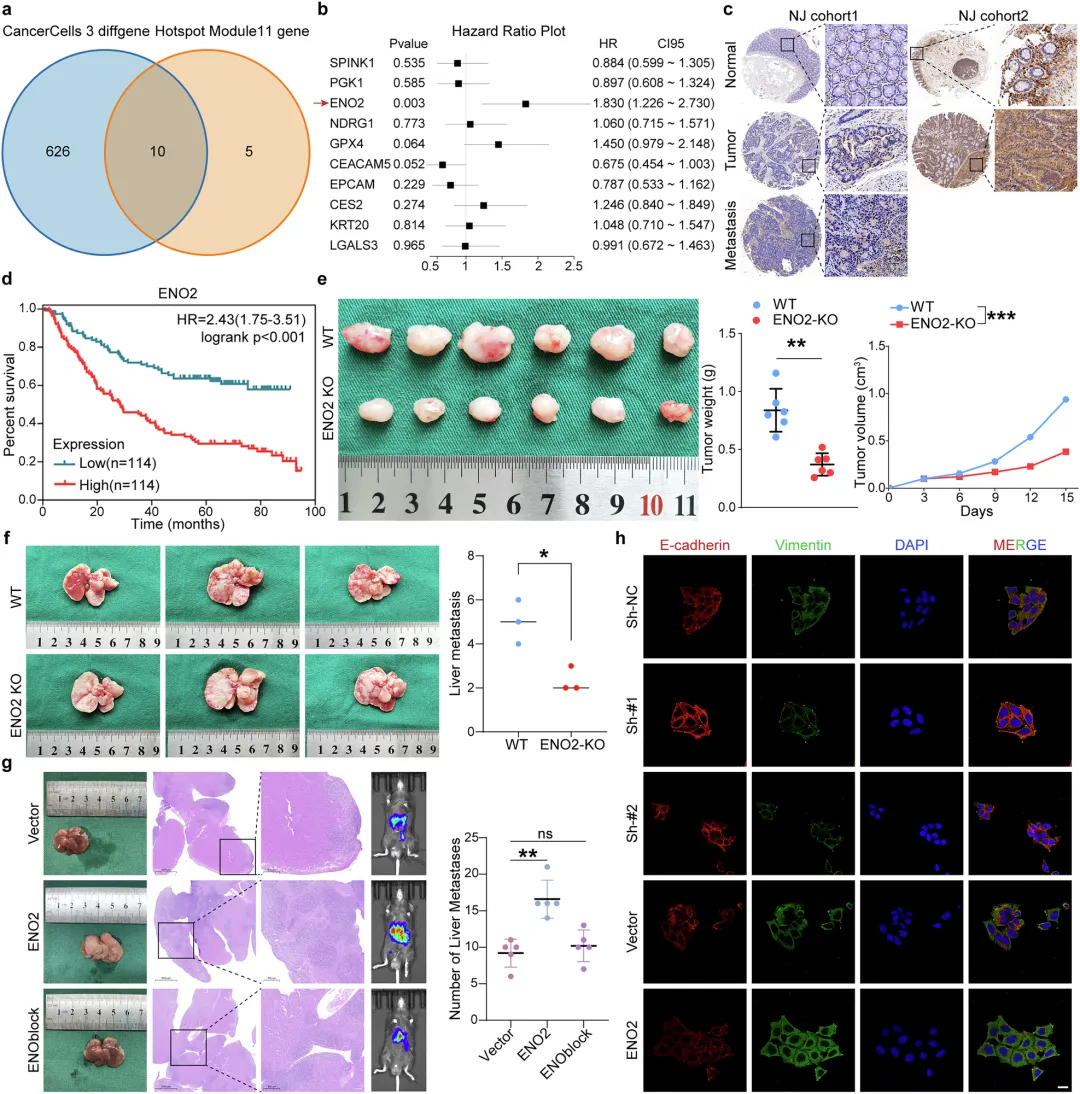
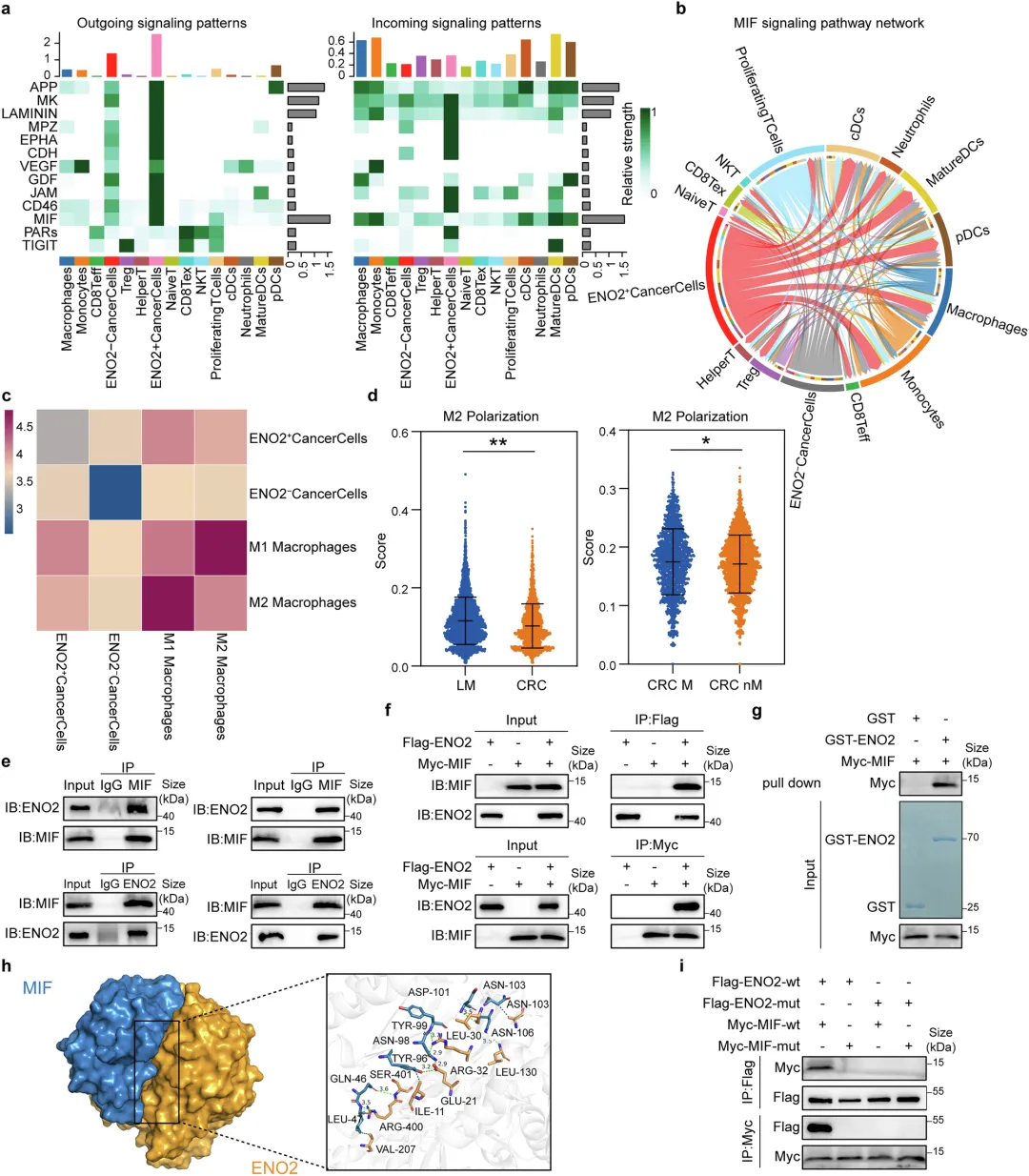
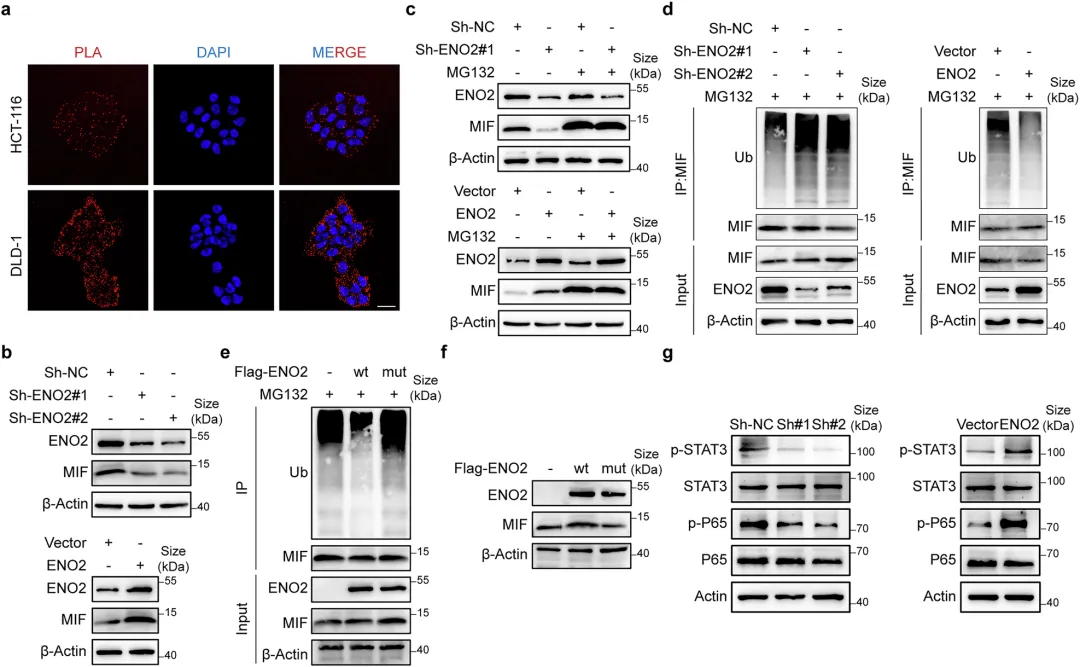
📌ENO2的“兼职”功能:不干活,当“保安”:ENO2本是糖酵解酶,但在CRLM中它“不务正业”——直接结合MIF蛋白,并招募HSP90形成三分子复合物,竞争性抑制E3泛素连接酶CHIP对MIF的K66位泛素化降解,从而稳定MIF。
📌MIF信号激活→M2极化→免疫抑制微环境:稳定后的MIF通过STAT3和NF-κB通路增强癌细胞自身恶性表型,同时通过旁分泌信号诱导肿瘤相关巨噬细胞向M2型极化,空间转录组证实ENO2⁺癌细胞与M2巨噬细胞在转移灶中高度共定位。
📌基因敲除 + 药理学抑制双验证:ENO2敲除的DLD-1细胞在小鼠脾脏注射模型中,肝转移结节减少50%以上;ENO2过表达则显著促进转移,且这一效应可被MIF抑制剂ISO-1或M2极化抑制剂LPS阻断。
结直肠癌肝转移是导致CRC患者死亡的最主要原因,5年生存率不足20%。肝转移灶的肿瘤微环境高度免疫抑制,M2型巨噬细胞大量浸润,是治疗失败的核心原因。但长期以来,一个关键问题悬而未决:究竟是什么“信号”从转移性癌细胞中发出,把本该清除肿瘤的免疫细胞“策反”成了帮凶? 本研究首次揭示,ENO2通过一种非代谢的“兼职”功能,充当了这个“策反计划”的总指挥。
✅单细胞转录组测序:3例CRLM患者(原发灶+癌旁+肝转移)+ 3例非转移CRC患者(原发灶+癌旁),共15份样本,57,674个细胞,构建CRLM单细胞图谱。
✅模块分析 + 多因素Cox回归:从“Module 11”和“cancer cell 3”特征中筛选10个候选基因,最终锁定ENO2为唯一与预后显著相关的基因。
✅体内外功能验证:ENO2敲除/过表达的DLD-1/HCT-116细胞→皮下成瘤、脾脏注射肝转移模型;THP-1巨噬细胞共培养体系检测M1/M2标志物(CD86/CD206)。
✅分子机制:Co-IP + 质谱鉴定ENO2互作蛋白;GST pull-down验证直接结合;分子动力学模拟预测结合界面;泛素化实验定位K66为关键泛素化位点。
✅空间转录组:验证ENO2⁺癌细胞与M2巨噬细胞在转移灶中的共定位。
✅患者来源类器官:构建ENO2敲除/过表达的PDO,与巨噬细胞共培养验证M2极化。
🔷细胞组成特征
🔷肝转移癌细胞恶性特征
基因组层面:拷贝数变异(CNV)评分升高,基因组不稳定性显著增强。
基因表达:细胞增殖、缺氧应答相关基因表达明显上调。
细胞功能:相较于原发灶癌细胞,转移细胞的增殖能力、缺氧适应能力大幅提升。
🔷整体结论
单细胞图谱完整勾勒出结直肠癌肝转移的细胞生态,肝转移灶癌细胞的整体恶性程度显著高于原发灶。
🔷肿瘤细胞亚群初步分析
🔷原发灶癌细胞亚群鉴定
🔷总结
模块11可作为结直肠癌转移细胞的特征基因集;原发灶中携带EMT特征的3号细胞亚群,是肿瘤发生转移的始动细胞。
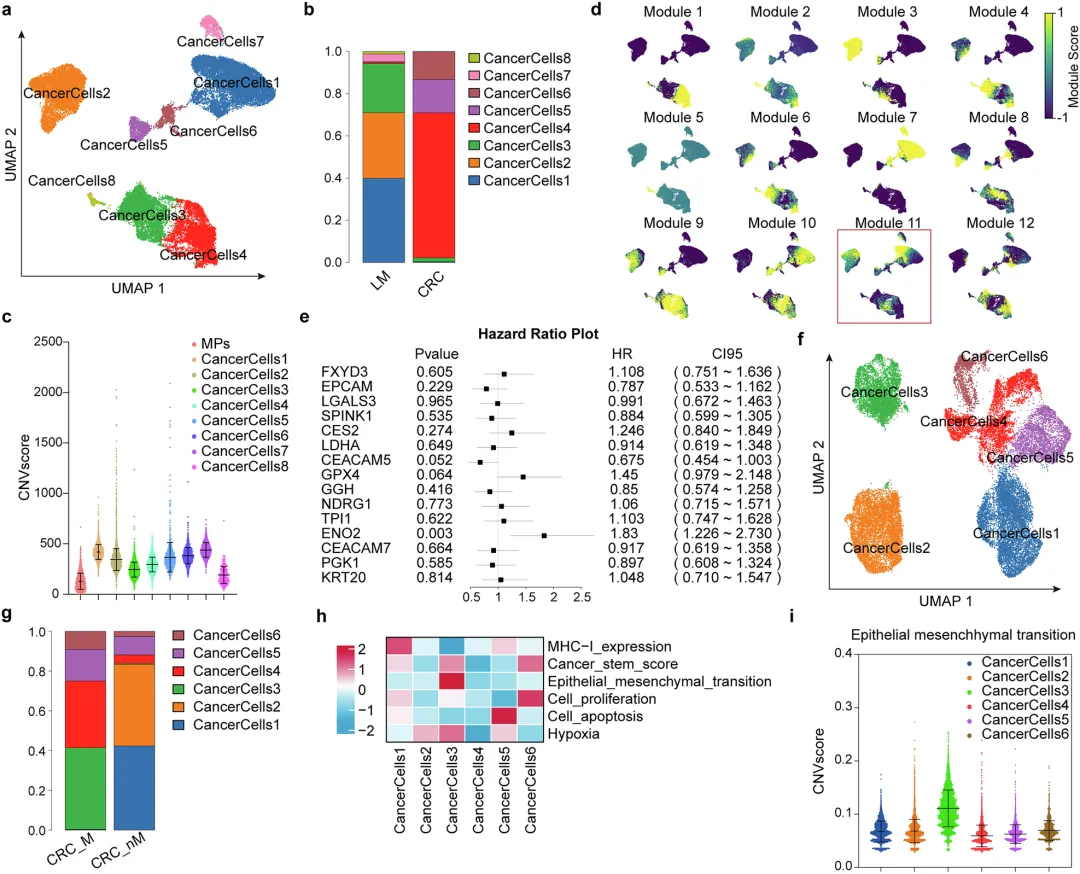
图2 基于模块的转移启动癌细胞亚群发现
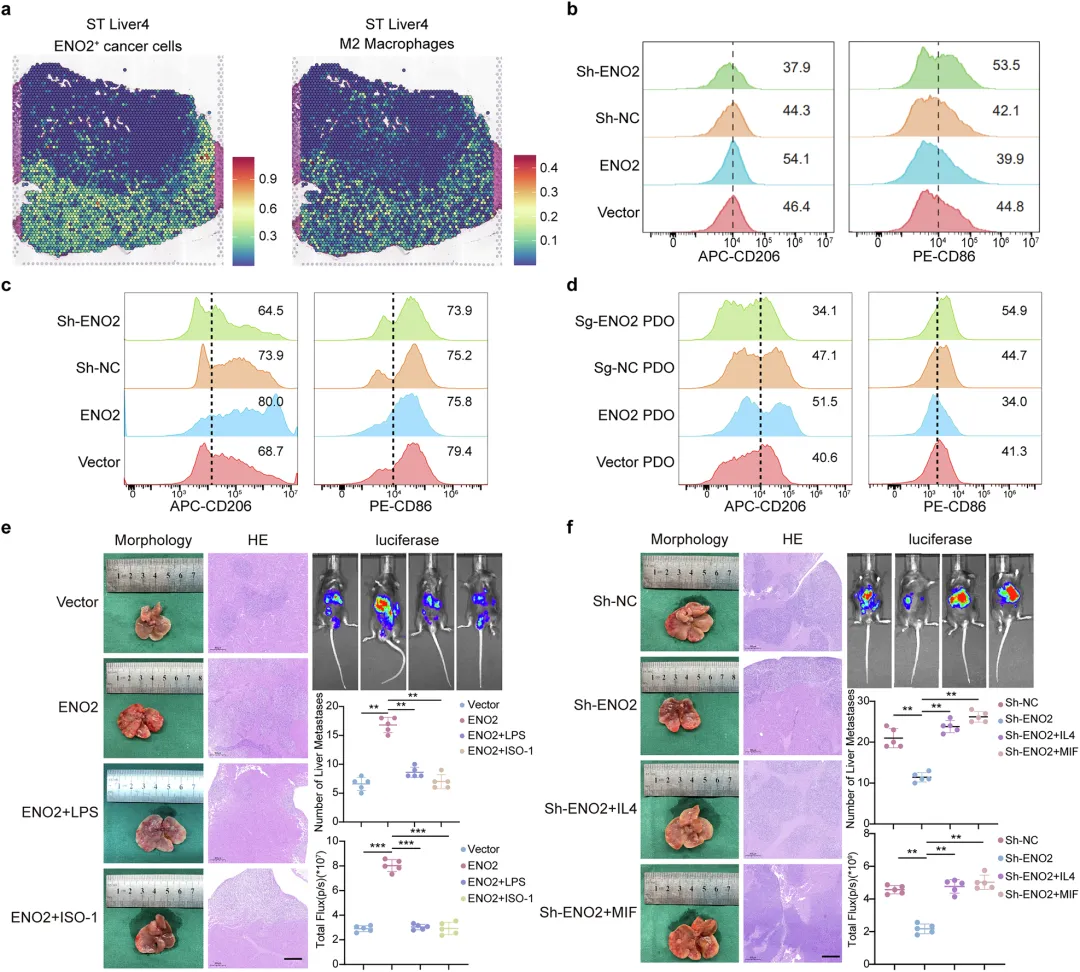
ENO2通过与MIF的直接相互作用来协调M2巨噬细胞的极化
🔷肿瘤微环境细胞通讯分析
利用CellChat分析细胞互作,ENO2⁺癌细胞是肿瘤微环境的核心信号枢纽,MIF通路信号传导十分活跃。
相比ENO2阴性细胞,ENO2阳性癌细胞与巨噬细胞的信号交流显著更强。
表型观测发现,转移灶巨噬细胞更易向促癌M2型极化,与生信分析结果吻合。
🔷蛋白互作关系验证
图4 ENO2通过与MIF的直接相互作用来协调M2巨噬细胞的极化
儿童癌症负担的预测变化