实现长寿命钠离子电池(SIB)的<15分钟快速充电技术仍是一项艰巨的挑战,这主要是由于硬碳(HC)界面处的副反应和不稳定的固态电解质界面(SEI)所致。
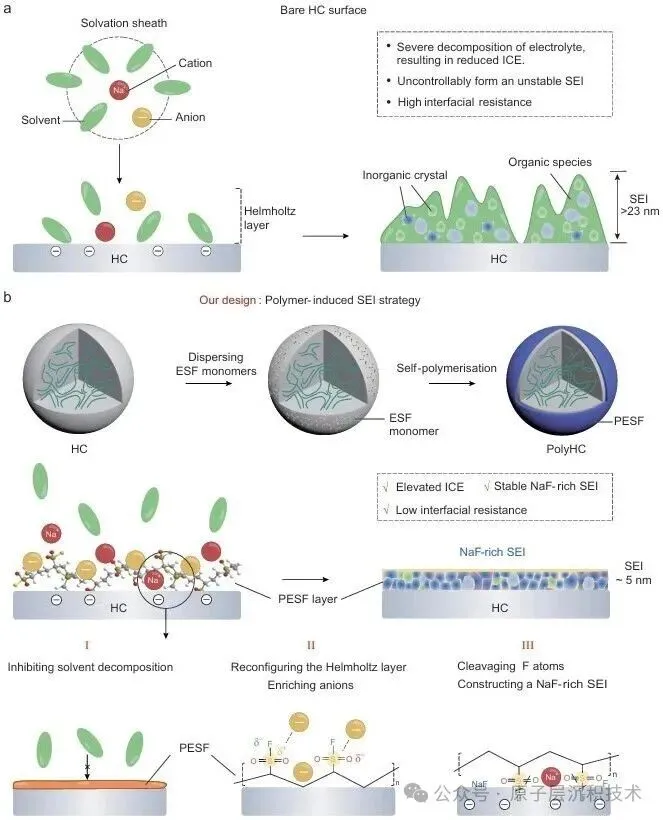
设计了一种<4.0 纳米的功能化聚合物分子层,即聚乙烯磺酰氟(PESF),涂覆在 HC 表面(PolyHC)上,以最大限度地减少电解质分解。带有-SO2F基团的PESF具有强大的极性特征,这同时诱导了PolyHC界面处的阴离子富集,并引入了额外的F原子,有助于形成约5.0 纳米的稳定SEI,该SEI结合了聚合物和NaF。这种具有弹性聚合物骨架的SEI永久性地固定了生成的无机成分,在快速充电过程中实现了长期的结构稳定性。
开发的这种通用的聚合物诱导SEI策略,使安时级SIB软包电池能够实现<10 分钟的快速充电能力。组装的1.2 Ah软包电池,搭配NaNi1/3Fe1/3Mn1/3O2正极和PolyHC负极,展现出卓越的快速充电能力和耐久性。该方法与多种氢化碳兼容,为调节氢化碳界面化学提供了新的视角。
为了提高市场竞争力,钠离子电池有望达到与锂离子电池相同的快速充电标准(美国先进电池联盟为锂离子电池设定了<15 分钟的快速充电时间目标)。然而,要同时实现<15 分钟的快速充电能力和长寿命,目前可用的钠离子电池仍面临巨大挑战。其中,HC特有的结构配置使其具备超过300 mAh/g的可逆钠存储容量,这一容量是由协同的“嵌入-吸附-纳米孔填充”机制所支持的。此外,碳层在钠化/脱钠过程中表现出的高度各向同性膨胀和收缩特性,赋予了HC极佳的结构稳定性,从而提高了循环稳定性和延长了使用寿命。然而,目前市场上可获得的HC仍然存在初始库仑效率低(ICE,大多低于90%)和快速充电能力差(充电时间超过30分钟)的问题,这严重阻碍了它们在实际超级离子电池中的应用。
HC/电解质界面化学,即决定固态电解质界面层(SEI)形成的化学过程,对这些性能限制起着关键的控制作用。与SEI形成密切相关的三个因素协同作用,限制了HC的快速充电能力:1)界面处的弱去溶剂化能力。界面处溶剂化结构中钠离子的缓慢逸出(去溶剂化过程)会显著影响硫化物离子电池的快速充电性能。2)界面处严重的副反应。电解质在氢氯化物界面处会失控分解,引发众多副反应,这会严重消耗电解质并使固体电解质界面变厚,从而增加不可逆的钠损失(低电解质离子浓度)和界面阻抗(较差的快速充电能力)。3)具有脆性和不稳定性的易破裂的SEI。在快速钠离子插入过程中,HC表面明显的体积变化和局部电场不均匀性加剧了SEI的断裂,使新的碳表面暴露于电解质分解中。这会导致SEI的持续增厚、界面阻抗增加以及严重的极化,最终削弱了快速充电能力和循环稳定性。
在商用HC表面上设计稳定、均匀、低阻抗的SEI仍然是一个重大挑战,但对于实现SIB的商业可行性范例至关重要。目前的主要策略包括预钠化处理方案、钝化涂层和先进的电解质工程。尽管预钠化化处理能提高商用HC阳极的离子传导效率,但这种处理方式无法同时促使电解质中形成稳定的SEI,同时还会带来极高的商业加工成本。此外,钝化涂层在HC/电解质界面实现物理隔离,从而减少副分解反应,并通过有策略地隔离电活性表面来提高离子传导效率。然而,这种技术所固有的复杂加工过程和高昂的经济成本从根本上限制了其商业可扩展性。先进的电解质工程促使在 HC 结合界面处优先分解阴离子,以生成富含无机成分的SEI,从而最大程度减少电解质分解,并构建出稳定的SEI。然而,要在复杂的电化学环境中实现HC结合界面处阴离子优先分解的精确协调仍是一项艰巨的科学挑战。迫切需要一种市场驱动的策略,既能提高商业HC的离子电化学稳定性,又能促进稳定的SEI形成——这对于加快具有15分钟以内快速充电能力的SIB商业化部署至关重要。
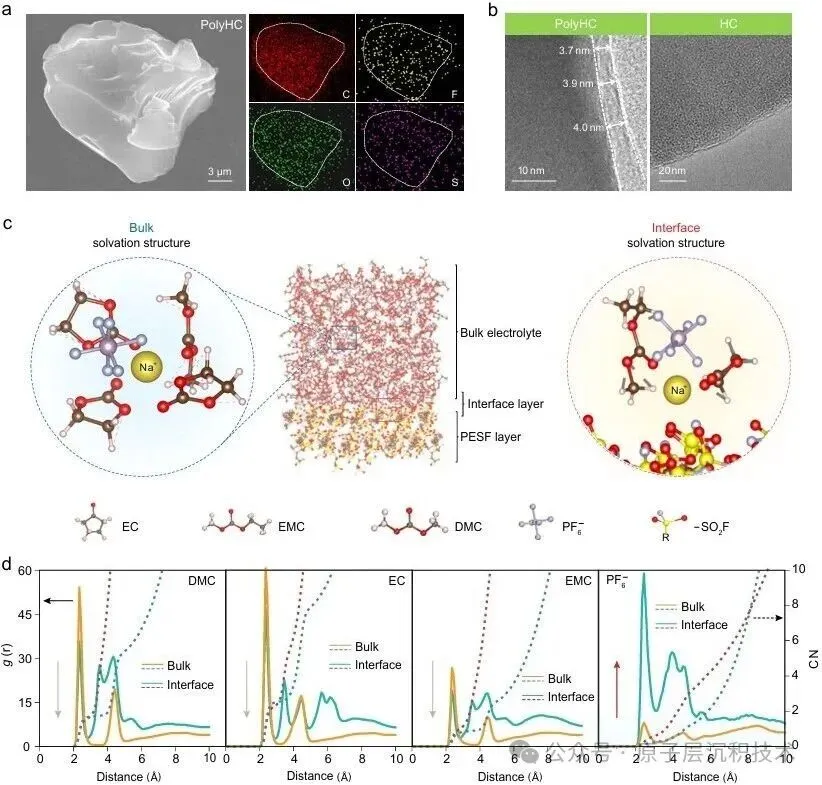
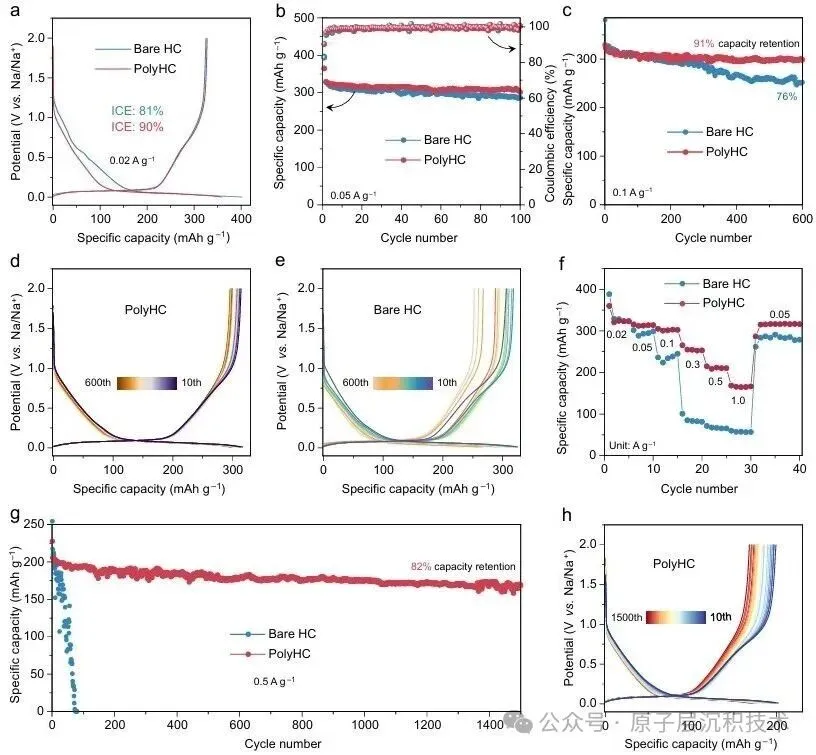
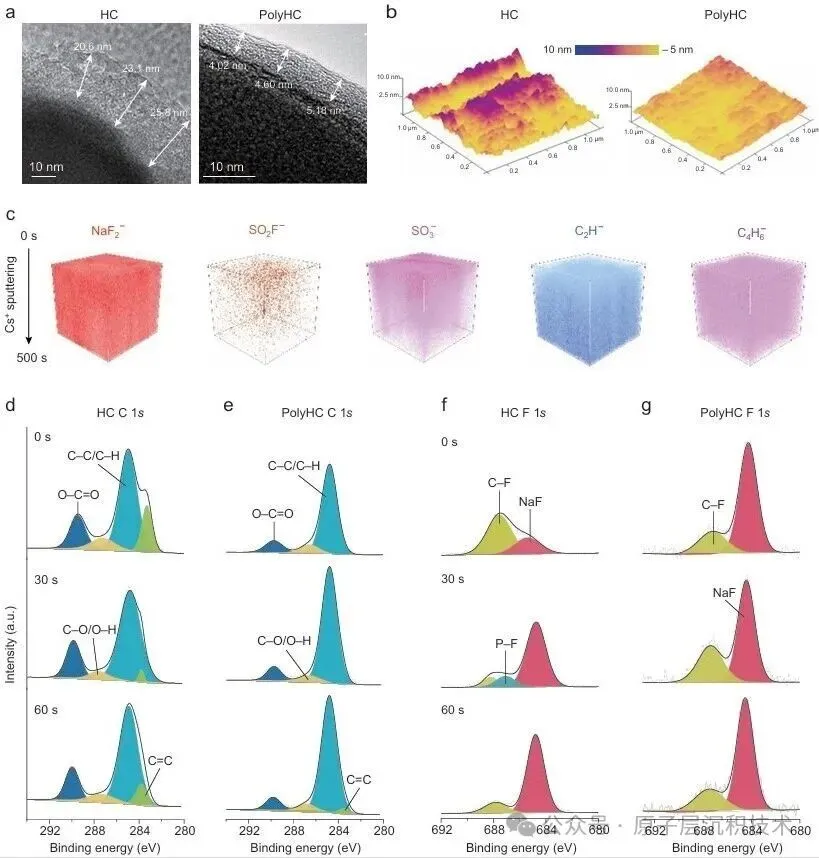
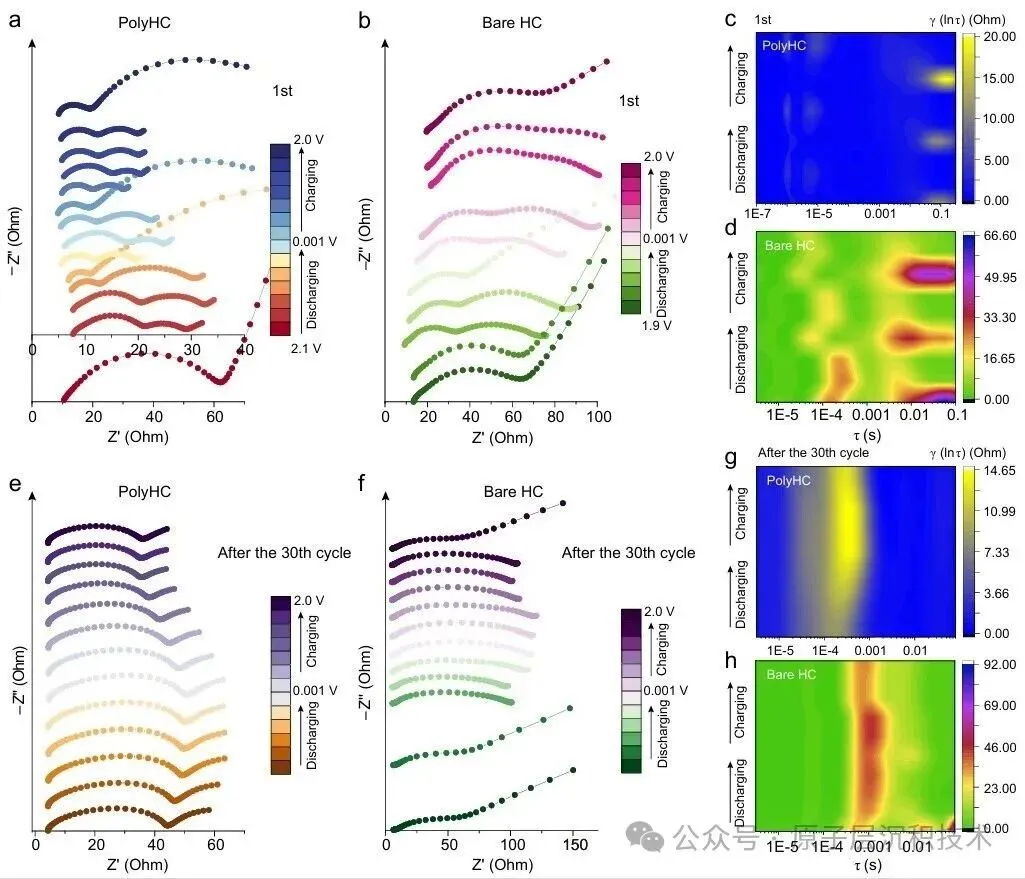
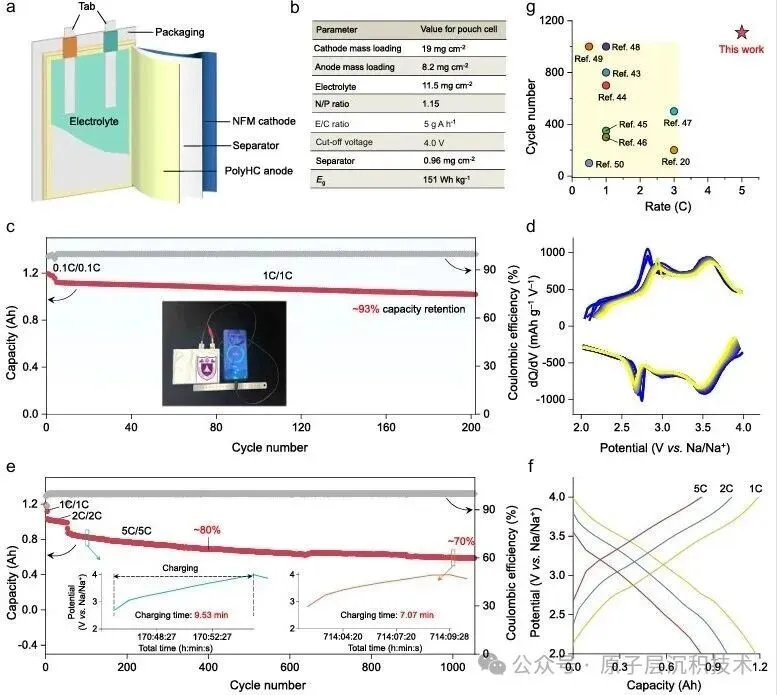
图1. 聚合物诱导SEI形成策略的设计。(a) 硬碳界面化学分析及其表面SEI模型。(b) PESF聚合物包覆硬碳的制备工艺及PolyHC表面的界面化学。图2. PolyHC界面化学分析。(a) PolyHC的SEM及其表面元素分布;(b) PolyHC与纯硬碳表面的TEM图像;(c) 电解液在聚合物界面处的分子动力学模拟,包含体相和界面处的溶剂化结构;(d) 基于分子动力学模拟得到的电解液组分(溶剂分子及阴离子)的定量空间分布,该结果揭示了PolyHC界面与本体电解液区域存在显著的分布特征差异。图3. 电化学循环测试。(a) 纯硬碳与PolyHC在0.02 A/g电流密度下的GCD曲线;(b) 纯硬碳与PolyHC在0.05 A/g下100次循环测试结果;(c) 纯硬碳与PolyHC在0.1 A/g下的循环耐久性,(d, e) 其对应的GCD曲线;(f) 纯硬碳与PolyHC的倍率性能;(g) 纯硬碳与PolyHC的长循环稳定性,(h) 及对应的GCD曲线。图4. 界面结构探测。(a) SEI形成后纯硬碳与PolyHC的TEM图像;(b) 经过数次循环后纯硬碳与PolyHC表面的AFM图像;(c) PolyHC循环后表面的飞行时间二次离子质谱(ToF-SIMS)检测结果;(d–g) 纯硬碳 (d, f) 与PolyHC (e, g) 循环后不同蚀刻深度下的XPS C 1s与F 1s特征峰。图5. PolyHC与bare HC界面动力学分析。(a, b) 两种材料在相同电流密度下的初始原位EIS曲线;(c, d) 拟合的原位分布弛豫时间(DRT)曲线;(e, f) 两种材料循环30次后的原位EIS曲线,(g, h) 及对应的DRT曲线。图6. 实用软包电池快充性能评估。(a) 匹配NFM正极与PolyHC负极的软包电池结构示意图;(b) 该软包电池的组件参数详情;(c) 软包电池在1C/1C倍率下的循环性能,以及 (d)其对应的微分容量曲线,(c) 中的插图为经该满电软包电池供电的商用手机实物图;(e) 软包电池在2C/2C与5C/5C倍率下的循环耐久性与稳定性测试结果,以及 (f) 其对应的GCD曲线,图 (e)中的插图为该软包电池在5C/5C快充模式下的时间-电压曲线;(g) 本研究成果与现有先进钠离子电池软包电池的快充性能对比。综上所述,开发了一种聚合物诱导SEI形成策略,可功能性提升商用硬碳的ICE与快充性能。所制备的PolyHC能够有效降低界面处电解液过度分解。PolyHC能诱导界面阴离子富集并提供额外氟原子,协同促成了~5.0 nm的聚合物基SEI。由此,PolyHC在酯类电解液中展现出90%的ICE,在0.5 A/g电流密度下可稳定循环1500圈,具备优异长循环性能,且在酯类与醚类电解液中均表现出卓越的倍率性能。组装的1.2 Ah钠离子软包全电池,实现了5C倍率快充能力,同时可稳定循环1000圈,彰显出极具吸引力的实际应用潜力。研究同时验证了该聚合物诱导SEI形成策略对多种商用硬碳具有适配性。该工作为设计钠离子电池快充硬碳负极提供了一种可行的界面构筑方案,并将为更精密的界面设计提供思路。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
