突破百年合成难题!南大团队实现无聚合物副产物的大环三聚体专一合成,登顶 Org. Lett.
在超分子化学与有机合成的交叉领域,大环分子一直是皇冠上的明珠 —— 从杯芳烃、葫芦脲、环三藜芦烃到柱芳烃,这些结构精巧的环状主体分子,撑起了主客体化学、分子识别、智能材料等一众前沿方向。但半个多世纪以来,一个核心困境始终困扰着全球化学家:环化与线性聚合的天生竞争,让目标大环总是伴随着环状同系物、长链聚合物等副产物,提纯难、收率低、选择性差,几乎成了大环合成的 “标配难题”。
就在近日,南京大学王乐勇、林晨、江菊研究员团队在国际顶级有机化学期刊《Organic Letters》发表突破性成果:以 1,2,3,4 - 四甲氧基苯与多聚甲醛为原料,通过动态 Friedel-Crafts 烷基化反应,完全抑制聚合副反应,专一性合成大环三聚体,实现了环化 - 聚合平衡的完美规避,为大环选择性合成开辟全新路径!
这篇题为An Exclusive Synthesis of Macrocyclic Trimer over Polymers的研究,不仅用 NMR 实时追踪、单晶衍射、DFT 计算全方位解析反应机理,更构建了一套仅生成线性低聚中间体→专一转化为目标大环的理想合成体系,彻底告别传统大环合成的副产物困扰!
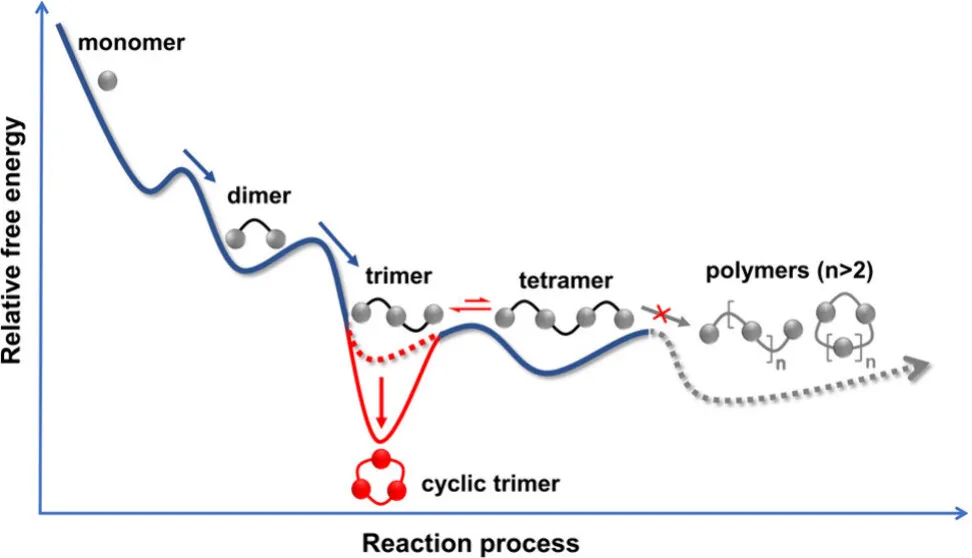
先给大家科普一个超分子化学的核心矛盾:芳香单体的缩合反应中,环化闭环与链增长聚合是一对天生对手。
过去几十年,无论是经典杯芳烃、热门柱芳烃,还是新型芳烃大环,合成路径几乎都逃不开图 1a 的宿命:
单体缩合时,既会发生分子内环化生成目标大环,也会持续链增长生成线性聚合物;
同时还会伴随不同聚合度的环状同系物(如环二体、环四体、环五体等);
副产物与目标产物结构相似、极性相近,分离提纯极度困难,目标大环收率大幅降低;
绝大多数体系需要模板辅助、预功能化、精密调控溶剂 / 催化剂等复杂策略,才能勉强提升选择性。
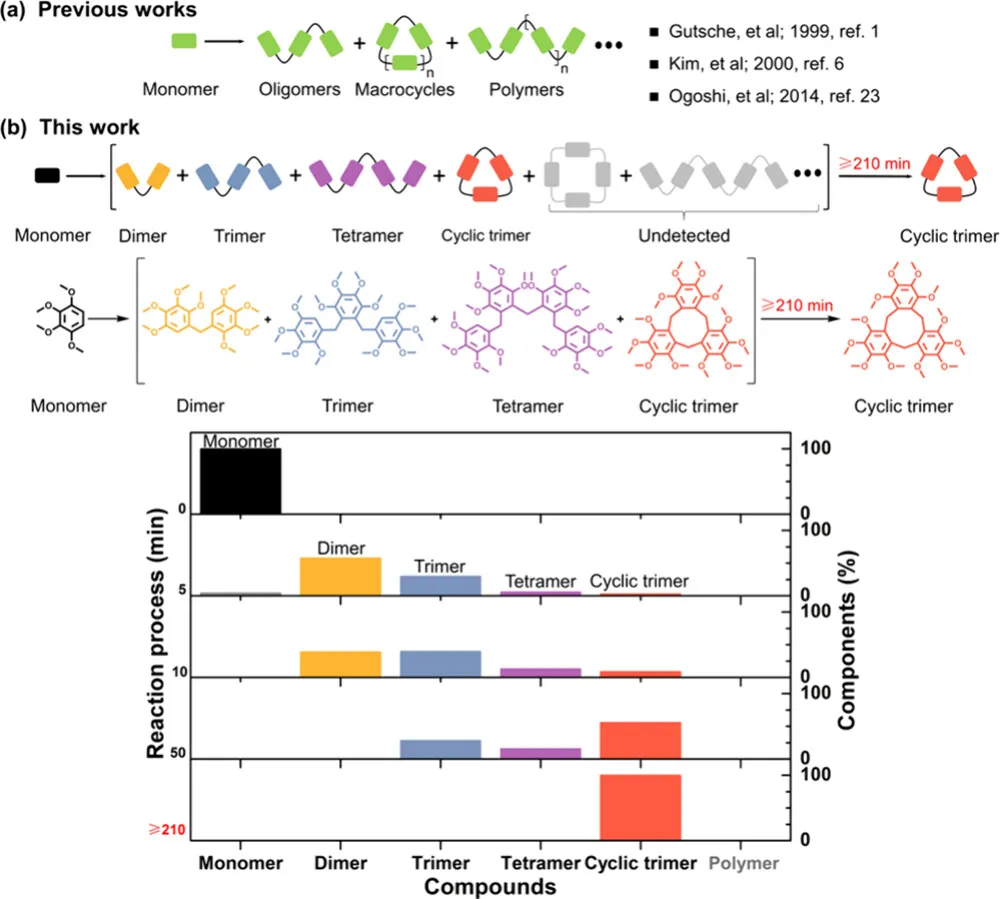
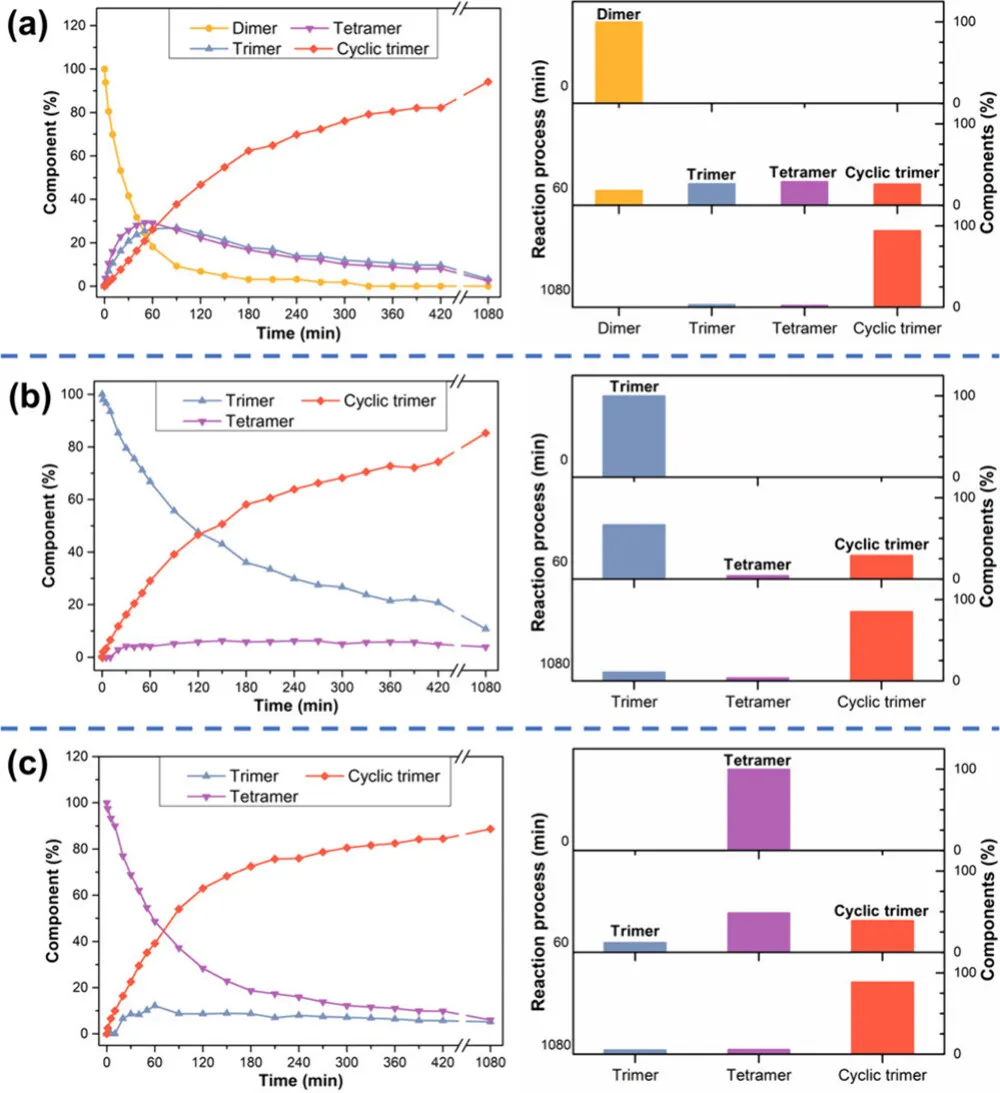
更棘手的是,学界对目标大环、环状同系物、聚合物三者的竞争机制缺乏系统研究,多数合成只能 “试条件”,难以从机理层面实现精准控制。而团队此次构建的合成体系,直接打破这一僵局 ——全程无聚合物、无环状同系物,仅生成线性二聚体、三聚体、四聚体中间体,最终 100% 转化为单一大环三聚体(图 1b),实现了大环合成的 “完美选择性”!
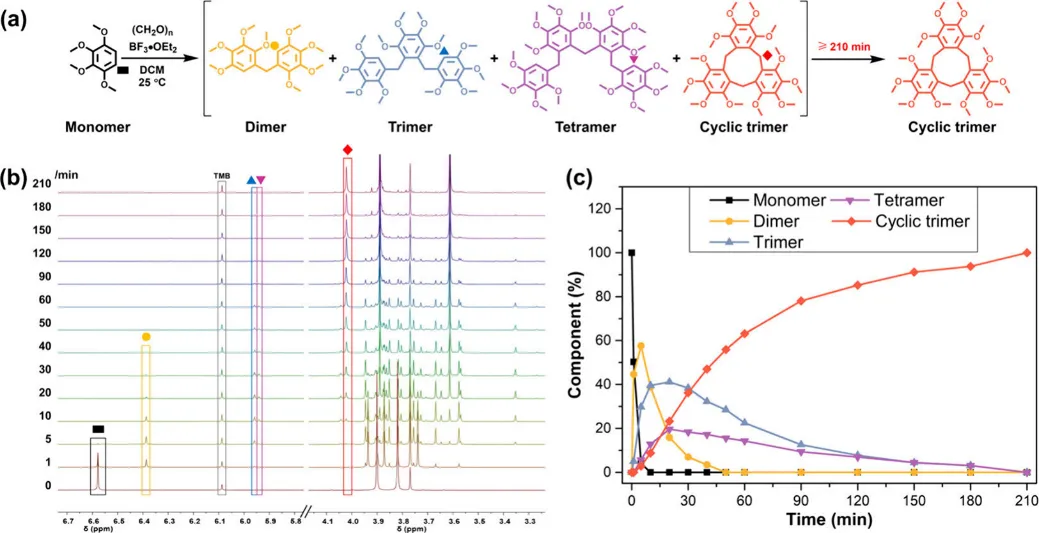
团队以1,2,3,4 - 四甲氧基苯与多聚甲醛为原料,二氯甲烷为溶剂,BF₃・OEt₂为催化剂,25℃常温下开展缩合反应,通过薄层色谱 TLC、时间分辨 ¹H NMR、单晶 X 射线衍射、高分辨质谱等手段,完整还原了反应全过程。
1. 反应进程:中间体逐步消失,大环独占终点
反应初期(15 min):单体快速消耗,生成 ** 线性二聚体(7.2%)、线性三聚体(42.1%)、线性四聚体(30.0%)、环状三聚体(14.4%)** 四种产物;
反应中期(50 min):单体完全转化,部分中间体逐步减少;
反应后期(210 min):所有线性中间体完全消失,体系仅存单一产物 —— 大环三聚体,相对含量达 100%;
全程监控:未检测到长链聚合物、未出现环二体 / 环四体等环状同系物,彻底杜绝传统副反应!
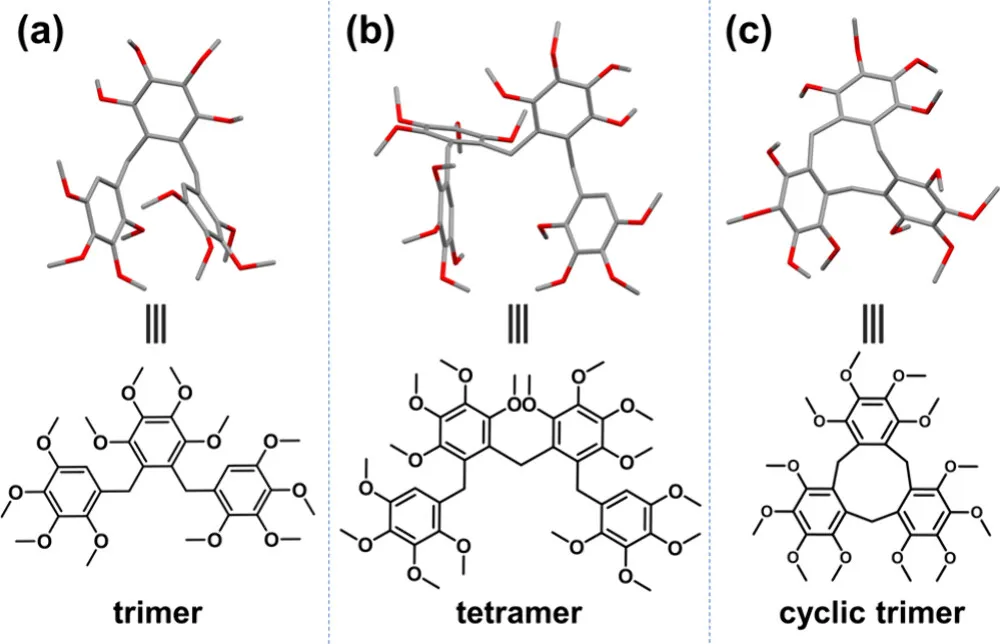
2. 结构确证:单晶衍射揭开构象奥秘
为了精准锁定产物结构,团队成功培养出线性三聚体、线性四聚体、大环三聚体的单晶,通过 X 射线衍射获得了原子级分辨率结构:
线性三聚体:呈现夹子型构象;
线性四聚体:呈现蜿蜒卷曲构象;
大环三聚体:因邻位甲氧基的空间位阻,形成独特扭曲构象,与 CTV(环三藜芦烃)衍生物结构类似,桥连亚甲基质子呈单峰,排除冠式构象可能。
3. 通用性验证:线性中间体均可专一转化为大环
团队进一步分离纯化线性二聚体、三聚体、四聚体,在完全相同的反应条件下单独反应,验证中间体的转化规律:
线性二聚体:先转化为线性三聚体、四聚体,最终 94% 生成大环三聚体,无高聚物;
线性三聚体:直接环化为主,少量解离重组,最终专一生成大环;
线性四聚体:通过断裂 - 重组路径,缓慢转化为大环三聚体;
大环三聚体:72 h 内无分解、无转化,热力学绝对稳定!
这一结果证实:所有线性低聚物都是活性中间体,而非副产物,最终都会定向奔赴热力学最稳定的大环三聚体。
三、机理解密:DFT 计算揭开 “专一选择性” 底层逻辑
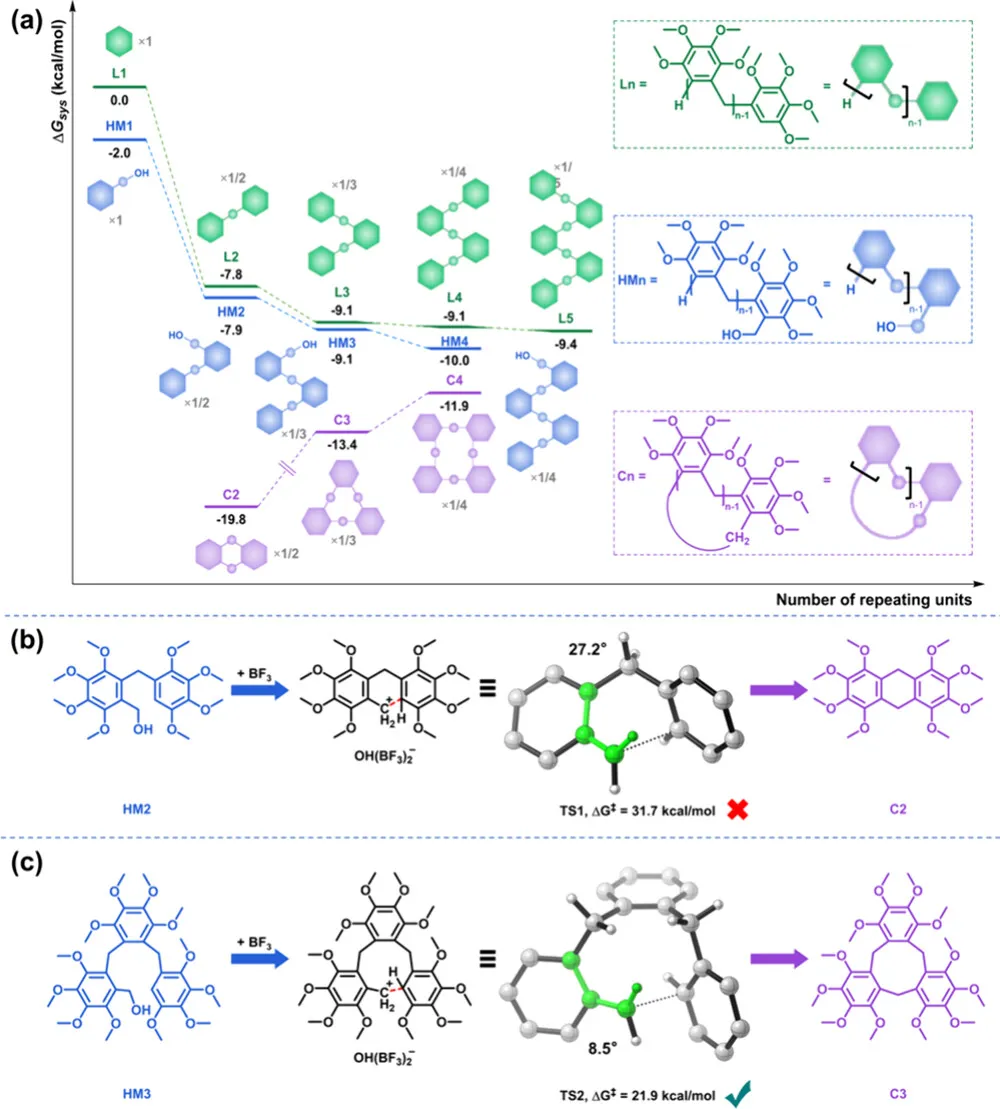
为什么这个体系能完全抑制聚合、只生成大环三聚体?团队通过密度泛函理论(DFT)计算,从热力学与动力学双维度,破解了这一核心问题。
1. 热力学:大环三聚体是体系最优解
反应体系为动态可逆 Friedel-Crafts 烷基化,最终会向体系总自由能最低的方向演化:
线性低聚物(L₂-L₄):随链长增加,焓变更负,但熵减显著,无法长期稳定;
羟甲基化中间体(HMₙ):能量低于线性低聚物,但动力学上快速转化,难以检测;
环状产物(Cₙ):环化比链增长多一步烷基化,熵损失更小,热力学稳定性远高于线性产物;
关键结论:大环三聚体(C₃)是所有可及产物中,热力学最稳定的物种。
2. 动力学:环二体被位阻 “卡死”,大环三聚体成唯一选择
热力学上最小的环二体(C₂)本应更稳定,但实验中完全未出现,根源在动力学能垒:
羟甲基二聚体(HM₂)→环二体(C₂):能垒高达31.7 kcal/mol,过渡态因环张力极度扭曲(C-C-C-H 二面角 27.2°),反应无法进行;
羟甲基三聚体(HM₃)→大环三聚体(C₃):能垒仅21.9 kcal/mol,与链增长能垒相当,环化顺畅发生;
四取代苯单体的大位阻效应,直接阻断长链聚合,同时卡死环二体路径,最终只通向大环三聚体!
这项发表于《Organic Letters》的工作,绝非一次简单的合成优化,而是从机理到应用,全面突破传统大环合成范式,其科学价值体现在三大维度:
1. 突破核心瓶颈:首次实现无模板、无副产物的大环专一合成
区别于传统需要模板、预功能化、精密条件调控的策略,该体系常温、常规溶剂、简单催化剂,即可实现 100% 选择性,彻底解决环化 - 聚合竞争的百年难题,为各类芳烃大环合成提供通用新思路。
2. 明晰竞争机理:建立 “中间体 - 产物” 的动态转化机制
通过 NMR 实时追踪与 DFT 计算,首次系统阐明线性低聚中间体→单一大环的分步转化路径,厘清热力学稳定性与动力学能垒的协同作用,填补了大环合成竞争机制的研究空白。
3. 拓展应用潜力:多修饰位点大环,赋能功能超分子材料
本次合成的大环三聚体拥有多个甲氧基修饰位点,可轻松实现构象固定、功能化衍生,在分子识别、传感、催化、智能响应材料等领域具备广阔应用前景,为新型超分子主体的开发提供优质骨架。
五、写在最后:从 “混合产物” 到 “专一合成”,超分子化学的又一次跨越
从 19 世纪杯芳烃的偶然发现,到 21 世纪柱芳烃的爆发式研究,大环分子的合成史,就是一部与副产物斗争、向选择性迈进的历史。
南京大学团队此次的突破,用一套简洁高效的反应体系,实现了 **“单体→线性中间体→纯大环三聚体”** 的完美闭环,让大环合成告别 “提纯焦虑”,走向精准可控。这不仅是有机合成方法学的重要进步,更为超分子化学、材料科学、生物医药等交叉领域,提供了更易得、更可控的大环分子平台。
正如论文结论所言,该体系完全规避环化 - 聚合平衡,为理解大环合成中的竞争机制提供了全新视角,而基于该大环的构象固定、功能修饰研究正在稳步推进,未来有望诞生更多高性能超分子功能材料。
DOI: 10.1021/acs.orglett.6c01048