期刊名:ACS Catalysis
DOI:10.1021/acscatal.6c01870 (2026)
羰基化反应是现代合成化学的基石,能够高效构建药物、农药与功能材料中普遍存在的羰基官能团,在有机合成领域占据着不可替代的核心地位。在已开发的羰基化催化体系中,钯催化羰基化反应已发展得十分成熟,钯催化剂与一氧化碳具有良好的兼容性,能够在相对温和的条件下高效完成氧化加成、CO 插入与还原消除的完整催化循环。与之形成鲜明对比的是,镍催化尽管具备地球丰度高、成本低廉、反应性独特等显著优势,其羰基化反应的发展却长期严重滞后。该领域一个核心且延续数十年的障碍在于,低价态镍对一氧化碳具有极强的热力学结合倾向,会快速生成配位饱和、完全无催化活性的四羰基镍(Ni (CO)₄)物种,这一过程会将镍催化剂锁定在非活性的静止态,既抑制了芳基卤化物的氧化加成过程,也阻碍了 CO 在后续催化步骤中的有效参与,最终导致芳基溴化物的温和镍催化羰基化反应长期难以实现。
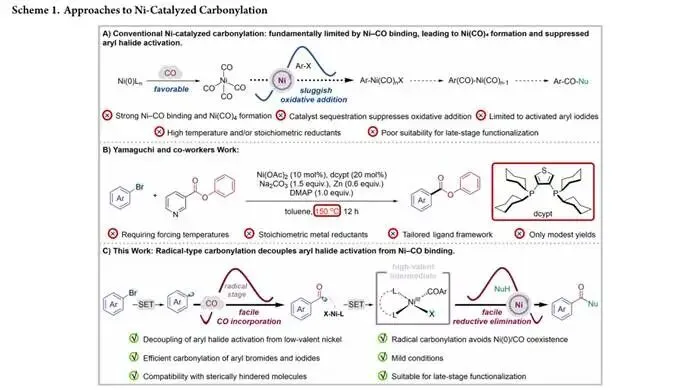
传统使用 CO 气体的镍催化羰基化反应,通常需要苛刻的高温高压条件、高度定制化的催化剂体系,且大多仅能适用于活性更高的芳基碘化物底物,芳基溴化物的镍催化羰基化更是极具挑战,在此前仅有山口团队的一例相关报道,该方法需要 150℃的高温、化学计量的金属还原剂与专属配体框架,且仅能得到中等的产物收率。为了缓解低价镍与 CO 之间的固有不兼容性,研究者们开发了一系列替代策略,包括使用 CO 替代物、化学计量镍配合物、基于转金属化的合成方法等,以此最大程度减少 Ni (0) 物种与 CO 的直接接触。尽管这些策略在一定程度上拓展了镍介导羰基化反应的范围,但它们大多依赖预官能化的芳基亲核试剂、较高的反应温度,或是仅适用于范围极窄的底物类型,始终未能为芳基卤化物、尤其是芳基溴化物的温和镍催化羰基化提供通用解决方案。与此同时,近年来光氧化还原催化领域的进展表明,通过自由基路径绕过高能的双电子反应步骤,能够突破过渡金属催化中长期存在的诸多局限,金属光氧化还原交叉偶联反应也已证实,动力学上难以发生的氧化加成过程,可被卤素攫取 / 自由基捕获的序列替代,从而实现传统双电子反应体系无法企及的反应性。受此启发,研究团队提出了全新的反应设计思路:能否通过解耦芳基卤化物活化与镍 - CO 结合过程,将羰基的引入步骤转移至自由基路径中,从而彻底避免 Ni (0) 物种与 CO 共存导致的催化剂失活问题。在这一设计中,光氧化还原条件下生成的芳基自由基会先发生自由基羰基化,再被镍物种捕获,从根源上杜绝了四羰基镍失活物种的生成,为芳基溴化物的温和镍催化羰基化提供了全新的解决方案。
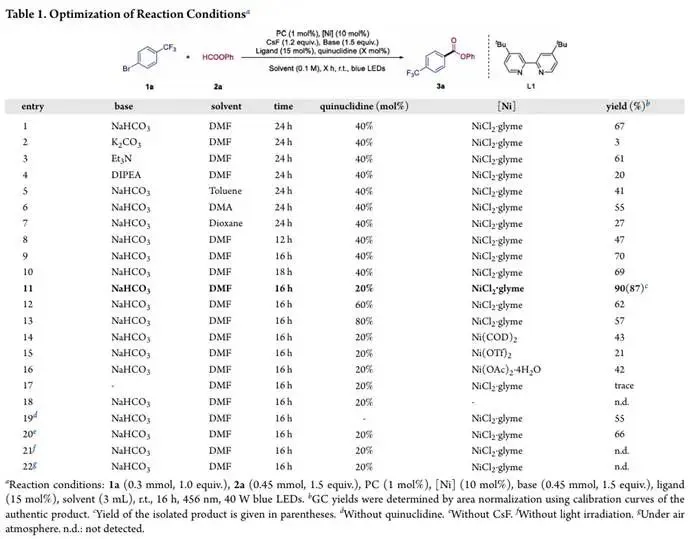
研究团队以对三氟甲基溴苯 1a 和甲酸苯酯 2a 为模型底物,对反应条件进行了系统的筛选与优化,最终确定了反应的最优条件:以456 nm 蓝光 LED 室温照射,无水 DMF 为溶剂,10 mol% 的 NiCl₂・glyme 为镍源,15 mol% 的4,4'- 二叔丁基 - 2,2'- 联吡啶为配体,1 mol% 的 4CzIPN 为光催化剂,1.5 当量的 NaHCO₃为碱,20 mol% 的奎宁环与 1.2 当量的 CsF 为添加剂,在该条件下,目标酯产物 3a 能以 90% 的 GC 收率和 87% 的分离收率获得。研究团队详细考察了各反应参数对反应结果的影响,在碱的筛选中,NaHCO₃表现出最优的效果,其他有机碱与无机碱均会导致反应效率大幅下降,这是因为乙酸根既能高效脱质子生成活性 NHC,也能作为质子穿梭体促进反应决速步的质子转移过程,实现了碱性与反应活性的完美平衡;溶剂效应同样十分显著,DMF 能给出最高的反应收率,甲苯、DMA 等溶剂仅能以较低效率生成目标产物;反应时间对收率呈现非线性影响,过长的光照时间会因催化剂或产物的光降解导致收率下降;奎宁环的负载量对反应至关重要,高于或低于最优的 20 mol% 都会导致收率显著降低;镍源的种类也显著影响反应效率,NiCl₂・glyme 的效果最佳,其他镍前体如 Ni (COD)₂、Ni (OAc)₂・4H₂O 的效果显著更差,Ni (OTf)₂则基本无反应活性。对照实验进一步证实,镍催化剂和碱是反应顺利进行的必要条件,省略任一成分都会完全抑制产物生成,而奎宁环和 CsF 虽能显著提升反应效率,但并非反应的必需组分,同时反应严格依赖光照和惰性气氛,在黑暗或空气条件下均无法检测到产物转化。
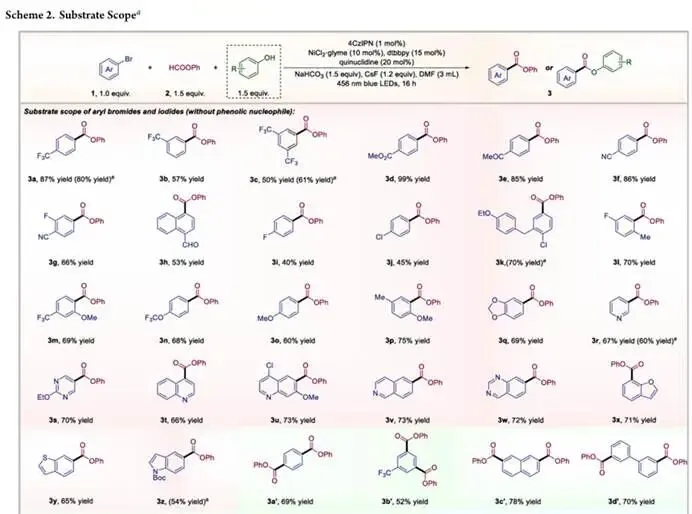
在最优反应条件下,研究团队对该反应的底物适用范围进行了全面考察, 该方法展现出极其宽泛的底物普适性和优异的官能团耐受性,首先考察了一系列电子效应多样化的单溴代芳烃底物,带有三氟甲基、酯基、酮基、氰基、醛基等强吸电子基团,以及氟、氯等卤素原子的底物,都能高效发生羰基化反应,烷氧基、甲基等给电子取代基也能很好地兼容,所有底物都能以中等至优异的收率得到对应的苯酯产物,其中敏感的醛基官能团能在反应中完整保留,充分体现了该催化体系的反应温和性。值得注意的是,该反应对底物的位阻效应极不敏感,邻位取代的大位阻底物也能高效转化,这一特性与自由基活化机制完全契合,成功规避了经典氧化加成和迁移插入步骤中典型的位阻限制。反应的底物范围还能顺利拓展到医药化学中极具价值的杂芳环体系,吡啶、喹啉、异喹啉、喹唑啉、苯并呋喃、苯并噻吩、吲哚等各类杂环底物,都能顺利发生羰基化反应,以良好收率得到目标产物,凸显了该方法在医药相关骨架合成中的广泛适用性。二溴代芳烃同样是合格的反应底物,能高效得到双羰基化的双官能团化产物,为分子的双位点修饰提供了有效方法。为了进一步证明该方法在复杂分子后期官能化中的实用性,研究团队考察了一系列结构复杂的生物活性分子衍生底物,包括维生素 E、雌酮、奥沙普秦、L - 薄荷醇、布洛芬、卡格列净关键中间体、L - 苯丙氨酸、尼泊金异丁酯、双丙酮果糖等天然产物和药物分子衍生底物,都能在标准条件下良好兼容,以 55%-72% 的合成有用收率得到目标羰基化产物,同时完整保留了复杂分子的原有结构与手性中心。除了芳基溴化物,芳基碘化物也是可行的反应底物,能以相当甚至更优的收率得到对应酯产物,同时酚组分的适用范围也得到了充分验证,各类取代苯酚都能很好地兼容,富电子和缺电子苯酚都能以中等至良好的收率得到对应的芳基酯产物。
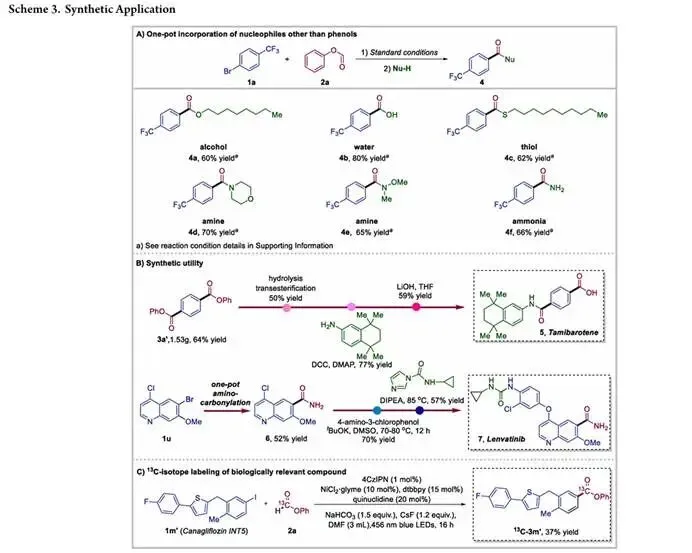
为了充分验证该羰基化方法的合成实用性,研究团队开展了克级制备、下游转化与合成应用研究, 该方法的核心转化是从芳基溴化物、甲酸苯酯和苯酚出发,一步实现芳基酯的羰基化合成,而对于醇、水、硫醇、胺、氨等其他亲核试剂,虽无法通过一锅单步反应直接引入,但可通过一锅两步的反应序列高效实现。研究团队以 1a 和 2a 为代表性底物,先在标准条件下完成羰基化生成芳基酯中间体,再原位加入稍过量的对应亲核试剂进行亲核取代,就能以稳定的良好收率得到烷基酯、羧酸、硫酯、酰胺等各类羧酸衍生物,极大地拓展了反应的合成应用范围。为了展示该羰基化平台在复杂分子合成中的价值,研究团队将该方法应用于药物分子高级中间体的简洁制备,克级规模的反应能顺利得到芳基酯产物 3a',收率 64%,后续经过水解、酯交换、胺化和最终水解,就能高效制备 FDA 批准的维 A 酸类药物他米巴罗汀;同时该方法还能通过一锅法操作,经后续的亲核醚化和氨基羰基化过程,高效合成抗肿瘤药物仑伐替尼的关键中间体 6,大幅缩短了该中间体的合成路线。除了复杂分子合成,该方法在分子工具开发中也展现出良好的效果,采用同位素标记的甲酸苯酯 (H¹³COOPh) 作为 CO 源,在标准催化条件下就能直接实现卡格列净中间体的位点特异性 ¹³C 标记,以 37% 的收率得到标记产物,充分证明了该羰基化方案与同位素标记策略的高度兼容性,凸显了其在药物代谢示踪研究和药物化学中的应用潜力。
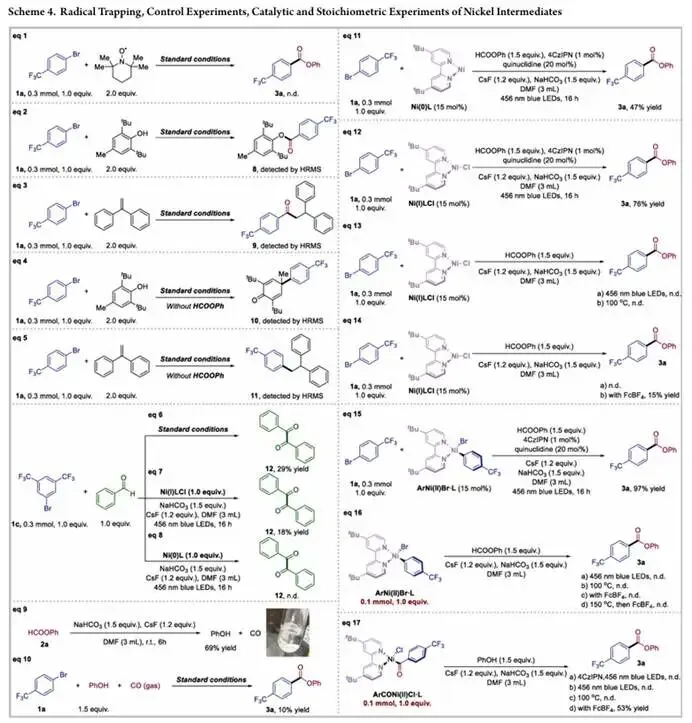
为了阐明反应的机理与高反应性的起源,研究团队通过自由基捕获实验、化学计量实验、光谱与动力学实验,对反应机理进行了全面深入的探究, 研究团队首先通过自由基捕获实验获得了自由基路径的直接证据,向标准反应体系中加入化学计量的自由基捕获剂 TEMPO,产物的生成被完全抑制;加入 BHT 或 1,2 - 二苯乙烯时,高分辨质谱能检测到对应的自由基加合物 8 和 9,与 CO 插入后生成的苯甲酰自由基中间体的捕获结果完全一致;而在无甲酸苯酯的条件下,加入 BHT 或 1,2 - 二苯乙烯则能捕获到羰基化之前的芳基自由基中间体,得到加合物 10 和 11,直接证明了芳基自由基的生成早于羰基的引入过程。以苯甲醛作为自由基探针的对照实验也进一步支持了芳基自由基中间体的参与,同时,碱促进的甲酸苯酯分解实验证实,甲酸苯酯会在碱作用下分解释放 CO 和苯酚,而在 CO 气氛下以苯酚替代甲酸苯酯仅能以 10% 收率得到目标产物,证明 CO 是反应的活性羰基源。
研究团队进一步通过定义明确的镍配合物开展了化学计量和催化实验,探究了镍在反应中的作用与价态变化。以 Ni (0) L、Ni (I) L、ArNi (II) Br・L 替代标准催化体系,分别能以 47%、76%、97% 的收率得到产物 3a,证明这些物种都是催化循环中的活性中间体;而在省略光催化剂和奎宁环的条件下,以 Ni (I) L 为唯一镍源,无论光照还是加热都无法生成产物,只有加入外源性氧化剂FcBF₄后,才能以 15% 的收率得到产物,说明虽然 Ni (I) 物种能与酰基自由基结合,但催化循环的顺利周转需要额外的氧化过程,以获得能实现后续成键步骤的高价镍物种。化学计量实验还排除了传统的闭壳层反应路径,分离得到的 ArNi (II) Br・L 配合物在光化学、热化学或氧化条件下都无法发生反应,即便加热到 150℃也无产物生成,不支持传统的氧化加成 - CO 配位 - 迁移插入的双电子反应路径;而分离得到的酰基 Ni (II) 配合物与苯酚和碱在光或热条件下都无法生成产物,只有经 FcBF₄氧化后才能顺利发生 C (O)-O键形成,以 53% 收率得到产物 3a,证明需要高价镍中间体(最可能为 Ni (III))来促进原本难以发生的 C (O)-O 还原消除步骤。
后续的光谱和动力学实验为反应机理提供了补充证据,¹³C 动力学同位素效应研究显示,芳基 C-Br 键的断裂是反应的决速步骤;Stern-Volmer 荧光淬灭实验表明,激发态的光催化剂 4CzIPN能被 Ni (dtbbpy) Cl₂、奎宁环和 NiCl₂・glyme 有效淬灭,循环伏安测试也证实了光激发的 4CzIPN对奎宁环的氧化在热力学上是可行的,其中 Ni (II) 物种对激发态光催化剂的氧化淬灭是主要反应路径,奎宁环的还原淬灭是辅助反应路径。灯光开关实验证实,反应需要持续的光照才能顺利进行,极低的量子产率也与自由基链机制不符,支持光氧化还原驱动的催化过程;原位红外光谱分析在 1900-2100 cm⁻¹ 范围内未检测到四羰基镍的特征吸收峰,证明该配位饱和的失活物种在反应条件下并未生成,从根本上验证了反应设计的合理性。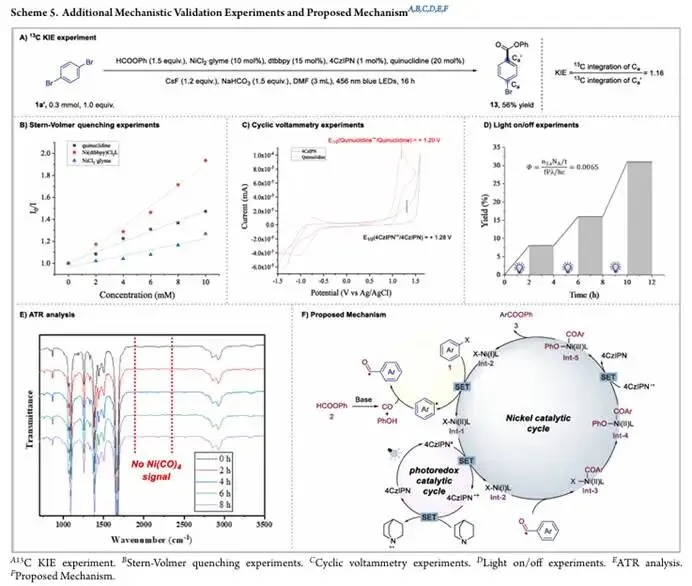
基于以上所有实验结果,研究团队提出了合理的协同自由基 / 高价镍双催化循环机理:光激发下,4CzIPN 被激发到激发态,与 Ni (II) 配合物 Int-1 发生单电子转移,生成 Ni (I) 物种 Int-2 和 4CzIPN 自由基阳离子;Ni (I) 中间体与芳基溴底物发生单电子转移,再生 Ni (II) 配合物 Int-1 并释放芳基自由基;与此同时,甲酸苯酯在碱促进下分解释放一氧化碳和苯酚;芳基自由基被 CO 捕获生成酰基自由基,再被 Ni (I) 配合物 Int-2 拦截得到酰基 Ni (II) 中间体 Int-3;苯酚与该中间体配位后生成 Int-4,再被 4CzIPN 自由基阳离子单电子氧化,生成高价 Ni (III) 中间体 Int-5,最终经还原消除得到目标酯产物,同时再生 Ni (I) 催化剂 Int-2,完成镍和光氧化还原双催化循环。

