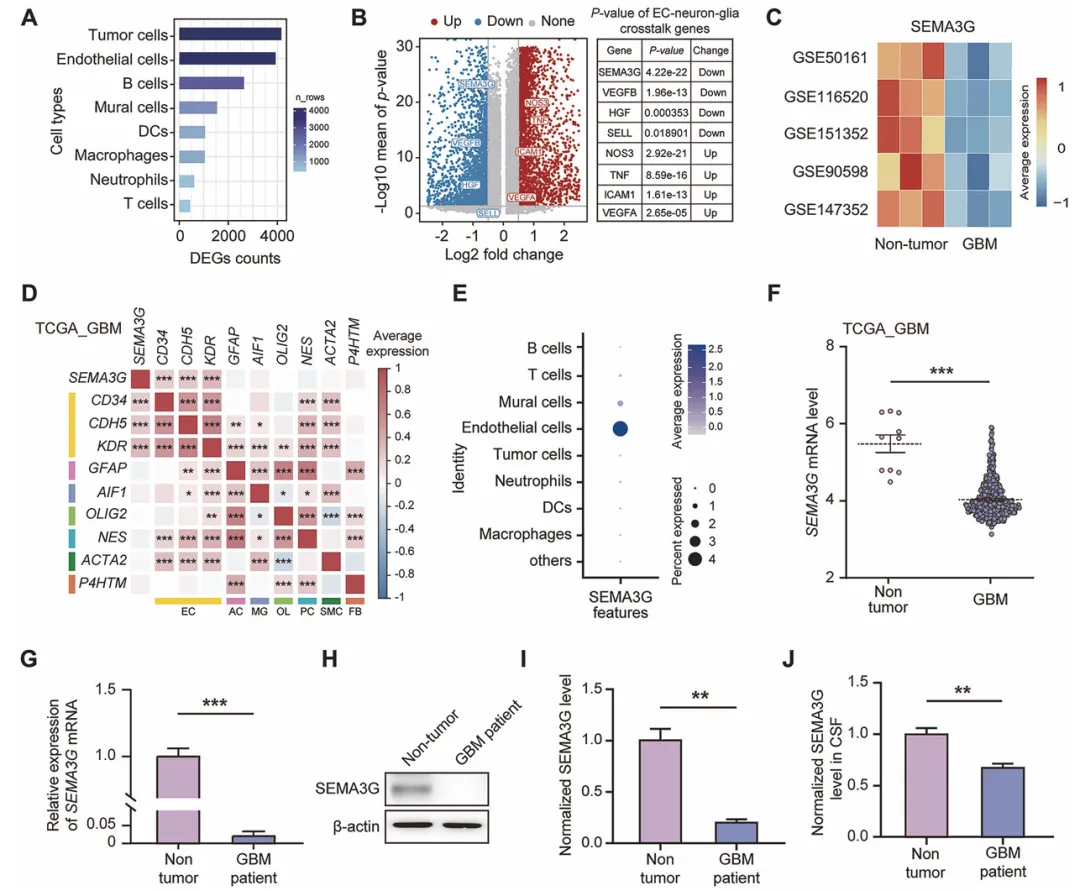
拿单细胞测序数据扫了一圈,发现SEMA3G在胶质母细胞瘤的血管内皮细胞里表达掉得特别狠,而且这个掉表达跟病人预后差有直接关系。他们还拿了病人的肿瘤组织和脑脊液来实测,发现不管是mRNA还是蛋白水平,SEMA3G都比正常脑组织少了一大截。
总之,SEMA3G在GBM里是低表达的,而且主要在血管内皮细胞里表达,这暗示它可能是个保护因子(图1)。
复现建议:去GEO或TCGA下载GBM的单细胞和bulk RNA-seq数据,用Seurat分析不同细胞类型的SEMA3G表达。再用自己收集的GBM和正常脑组织样本,做qPCR和Western Blot验证表达差异,顺便用ELISA测一下脑脊液里的SEMA3G浓度。
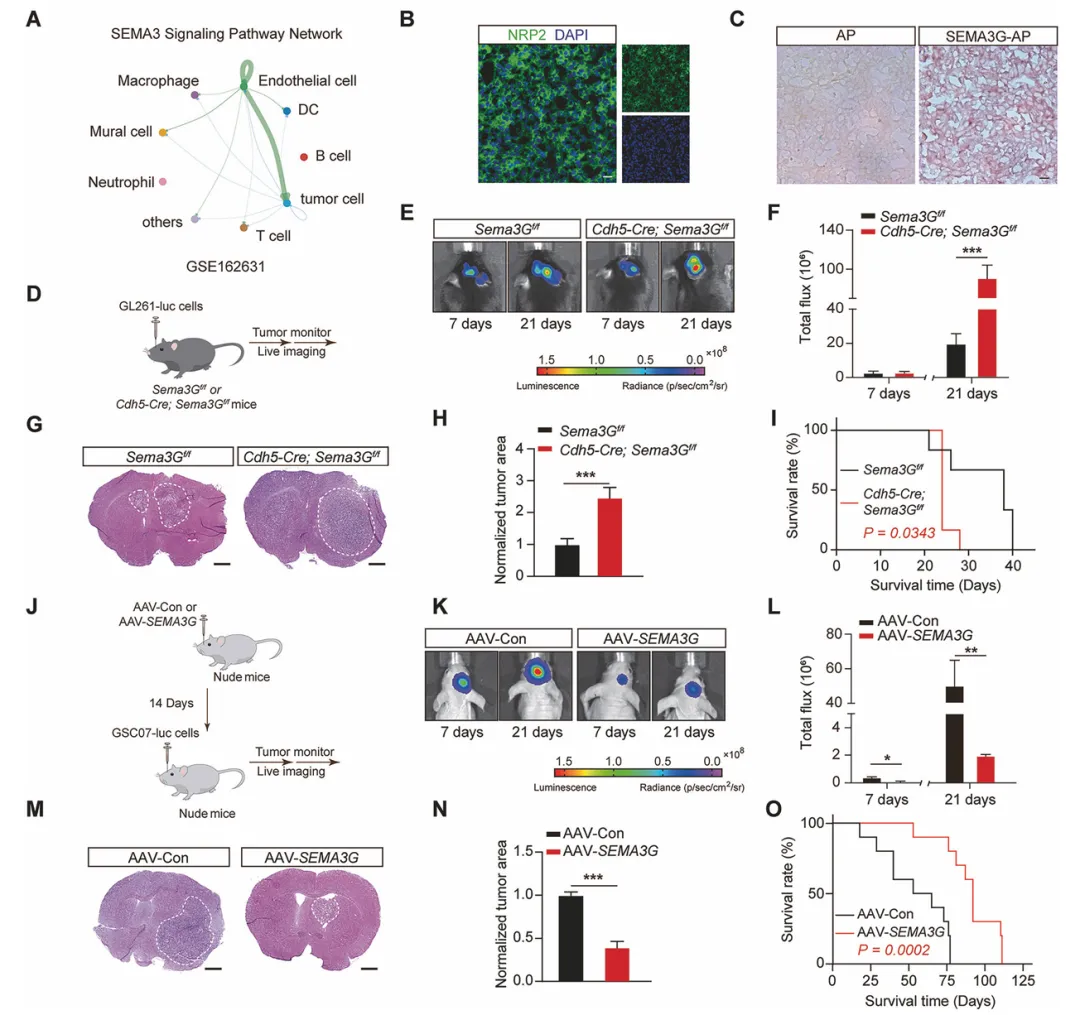
先用CellChat预测发现,SEMA3G的主要作用对象是肿瘤细胞。然后在小鼠脑子里特异性地敲掉内皮细胞的Sema3G基因,结果肿瘤长得飞快、老鼠死得也早。反过来,给老鼠脑子过表达SEMA3G或者直接打重组蛋白,肿瘤就明显变小了,老鼠活得更久。这就说明内皮来源的SEMA3G确实能压制GBM进展(图2)。
复现建议:用Cdh5-Cre工具鼠和Sema3G-floxed小鼠做内皮特异性敲除,原位注射GL261-Luc细胞后定期活体成像。过表达组用AAV-SEMA3G立体定位注射,两周后再种肿瘤。治疗组可以在种瘤后3天开始瘤内注射重组蛋白,每3天一次。
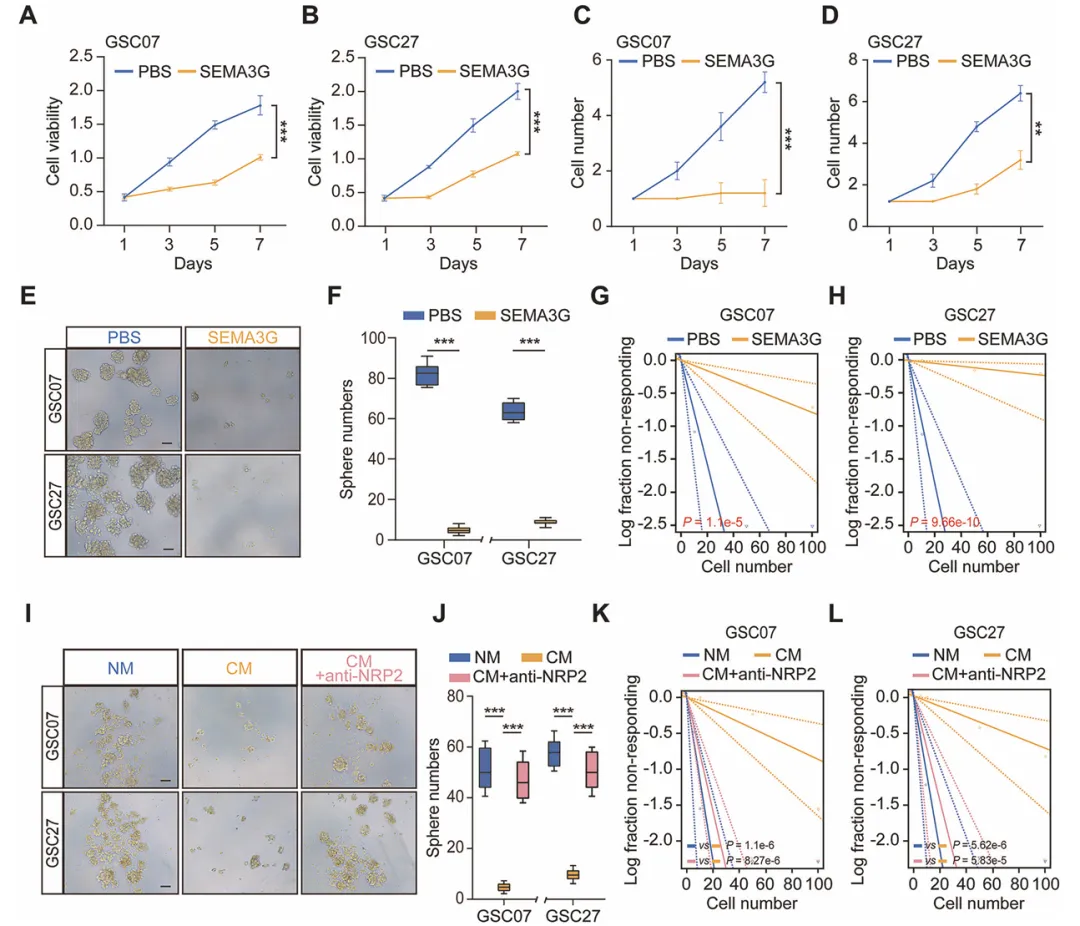
把重组SEMA3G蛋白加到GSC细胞里,发现细胞活力和成球能力都明显下降,极限稀释实验也显示干细胞比例降低了。他们还做了过表达SEMA3G的HUVEC,把这种细胞的上清加到GSC里,同样能抑制干性。但如果先用NRP2中和抗体处理,这种抑制效果就消失了,说明SEMA3G确实是靠NRP2起作用的(图3)。
复现建议:用慢病毒构建SEMA3G过表达的HUVEC,收上清做条件培养基。GSC细胞用Neurobasal+B27+EGF+bFGF培养。用CCK-8测增殖,极限稀释法算干细胞频率,成球实验拍照计数。NRP2中和抗体加进去做阻断,看效果是否被逆转。
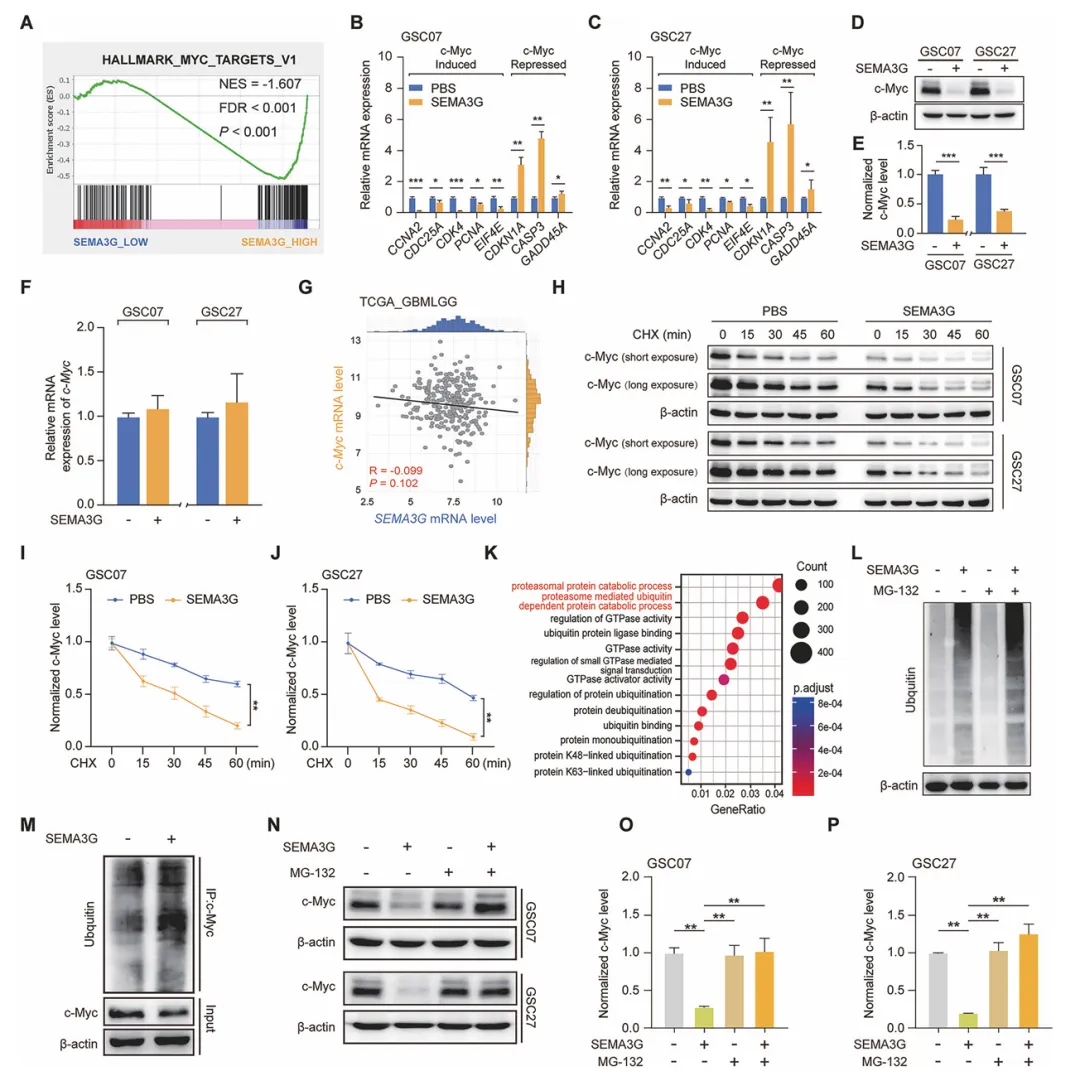
他们发现SEMA3G处理GSC后,c-Myc蛋白水平明显下降,但mRNA没变,说明是翻译后调控。放线菌酮实验显示c-Myc的半衰期明显变短了。再用MG132抑制蛋白酶体,c-Myc的下降就被完全逆转了。而且SEMA3G处理后c-Myc的泛素化水平显著升高。结论就是SEMA3G通过泛素-蛋白酶体途径让c-Myc降解(图4)。
图4 SEMA3G在GSCs中降低c-Myc蛋白的稳定性
复现建议:用重组SEMA3G处理GSC细胞48-72小时,收蛋白和RNA做Western Blot和qPCR检测c-Myc。CHX实验加CHX不同时间点收样。泛素化实验用MG132预处理6小时,再用co-IP富集c-Myc,泛素抗体检测。
先筛了一圈可能的E3连接酶,发现只有WWP2能和c-Myc结合。过表达WWP2会让c-Myc蛋白变少、泛素化增加、半衰期变短;沉默WWP2则相反。关键的是,如果先把WWP2敲低,SEMA3G对c-Myc的降解作用就完全没了,对GSC成球的抑制也减弱了。这就说明SEMA3G是靠着WWP2来降解c-Myc的(图5)。
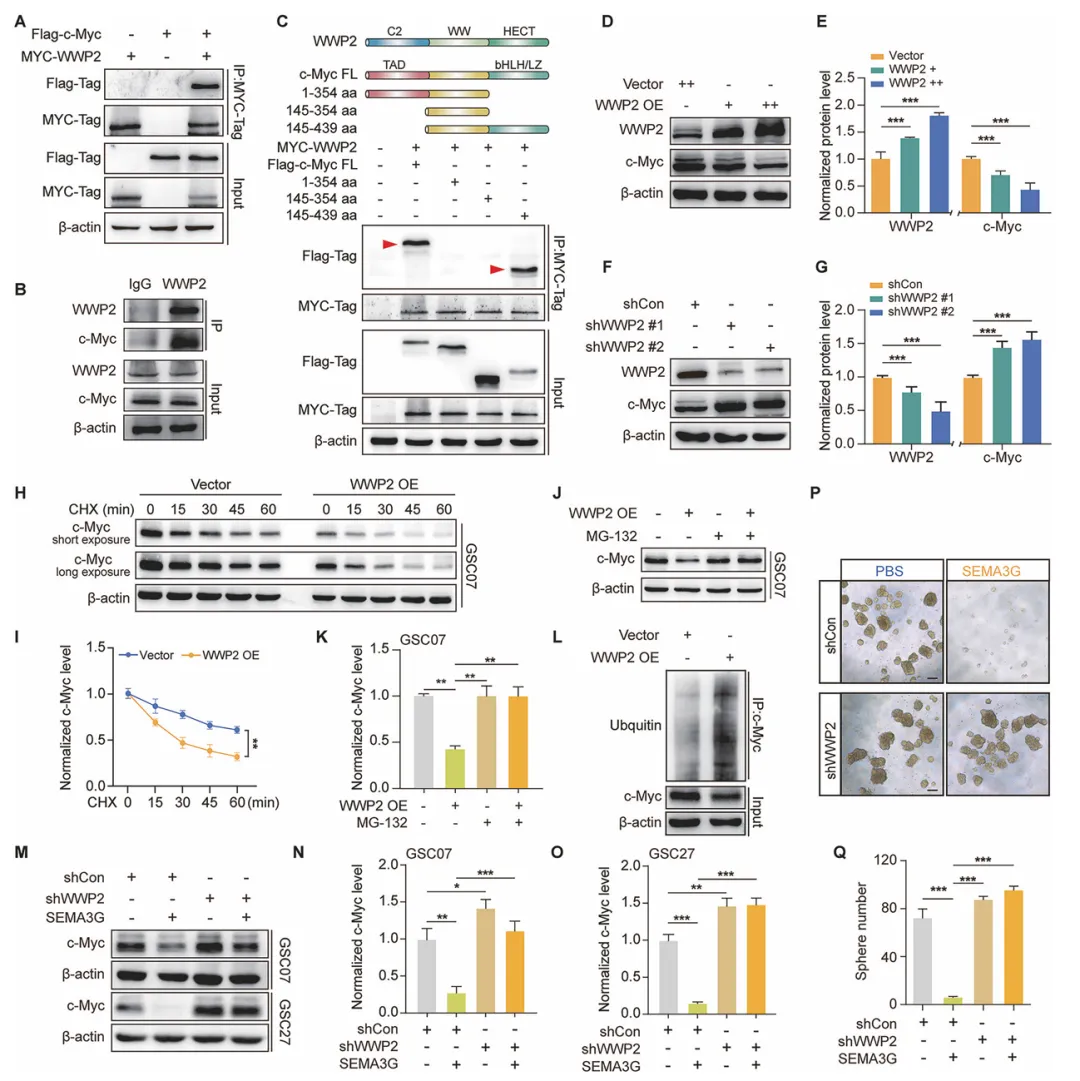 图5 SEMA3G通过依赖WWP2的方式促进c-Myc降解
图5 SEMA3G通过依赖WWP2的方式促进c-Myc降解
复现建议:用慢病毒shRNA做WWP2稳定敲低,或用过表达质粒做瞬时过表达。检测c-Myc用Western Blot,泛素化用co-IP。CHX实验测半衰期。成球实验看敲低WWP2后SEMA3G还能不能抑制成球。结合String数据库预测相互作用。
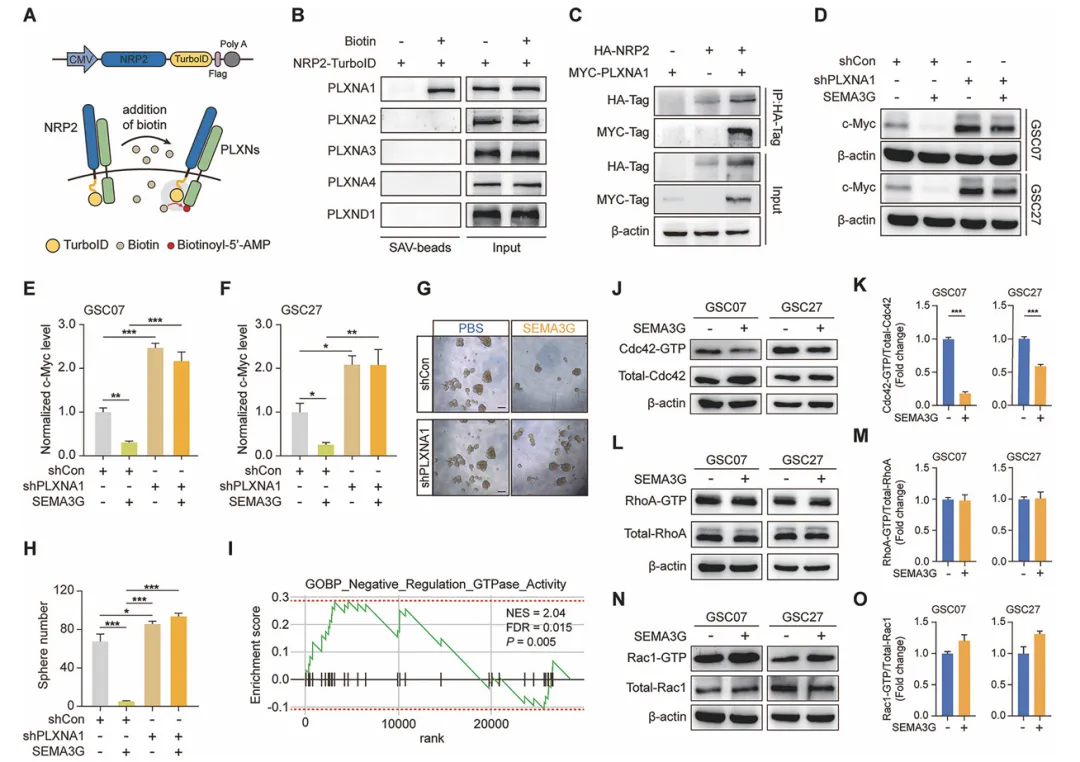
用TurboID邻近标记技术发现,SEMA3G的受体复合物是NRP2和PLXNA1。沉默PLXNA1之后,SEMA3G对c-Myc的降解作用就消失了。进一步分析发现,SEMA3G处理会显著降低Cdc42的活性,但不影响RhoA和Rac1。说明SEMA3G是通过NRP2/PLXNA1这条通路让Cdc42失活的(图6)。
图6 SEMA3G通过GSCs中的NRP2/PLXNA1失活Cdc42
复现建议:用shRNA敲低PLXNA1,再给SEMA3G处理,检测c-Myc变化。Cdc42活性用GST-CRIB pull-down检测。TurboID实验把NRP2和miniTurbo融合表达,加biotin标记,再用链霉亲和素磁珠富集,质谱鉴定互作蛋白。