陈德院士领衔!南京理工大学马宏飞/苏州纳米所崔义,最新Nature子刊!碳载体上界面Ru/RuOₓ异质结构调控木质素加氢脱氧选择性
- 2026-05-30 16:54:19


本研究针对木质素选择性转化制高值化学品与生物燃料的挑战,提出了一种基于碳纳米纤维(CNF)负载钌催化剂(Ru/CNF)的一锅法催化策略,实现了木质素富集玉米芯及真实木材生物质的高效氢解脱氧(HDO)转化。
通过构建界面Ru/RuOx异质结构活性位点,该催化剂在单步反应中实现了49.1%的液态烃质量收率,并高选择性地生成环烷烃。高温处理催化剂可调控产物分布,获得芳香化合物。放大实验证实了该工艺的可行性,收率达44%。该工作不仅为木质素定向转化为可持续燃料与氢载体提供了高效催化剂,也为通过催化剂界面工程设计调控生物质转化路径提供了重要借鉴。
要点1. 构建了具有明确异质界面的Ru/RuOₓ双功能活性位点
作者通过热诱导碳纳米纤维(CNF)表面羟基重构,成功构建了Ru/RuOₓ异质结构催化剂。这一界面结构形成了具有极化特性的Oδ⁻···Ruδ⁺···Ruδ⁺活性位点,能够协同异裂活化H₂并强烈极化酚类中间体的C-O键,从而显著降低加氢和脱氧反应的能垒,实现高效的一步法转化。
要点2. 阐明了通过调控金属-载体界面精准控制产物选择性的机制
研究发现,通过改变CNF载体的预处理温度,可以精确调控Ru/RuOₓ界面的氧物种含量,从而实现对最终产物选择性的“开关式”控制。富含氧的界面优先生成环烷烃,而去除氧物种后则转向生成芳烃,这为定向合成目标化学品提供了创新的催化剂设计原则。
要点3. 验证了催化剂在复杂真实生物质及放大条件下的稳健性
该催化剂不仅在模型化合物中表现优异,更在富含木质素的玉米芯乃至复杂的真实木质生物质原料中,均能实现完全脱氧并高选择性地生成液态环烷烃。放大五倍的实验取得了44%的质量收率,且催化剂在反应后未发生金属团聚或化学态变化,证明了其在实际应用中的可扩展性与稳定性。
图1 | 用于HDO反应的方法示意图
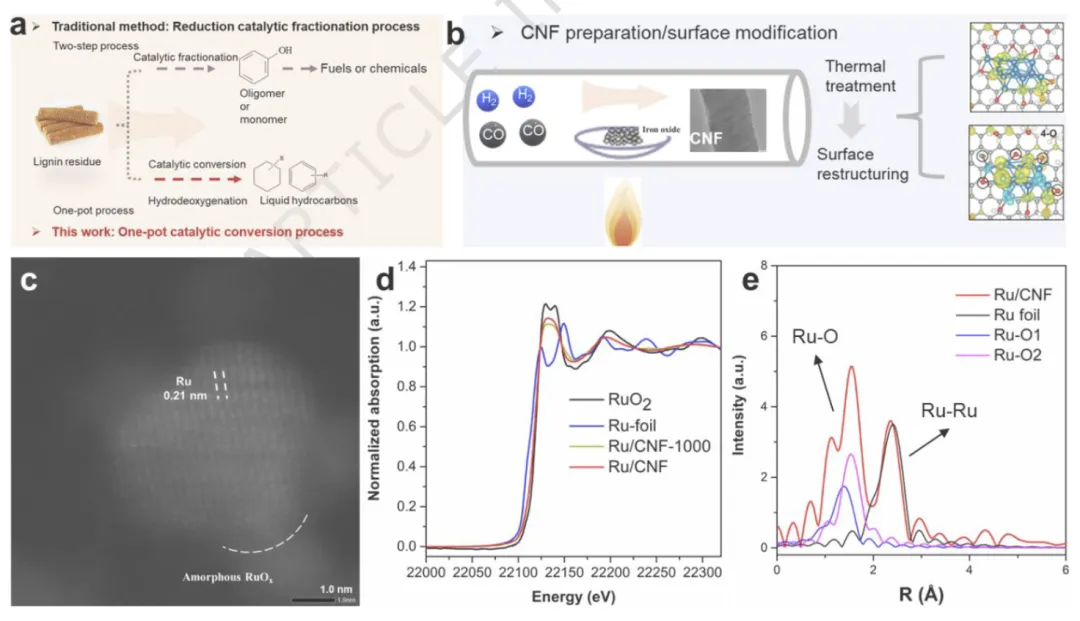
图1 用于HDO反应的方法示意图
(a) 木质素转化的传统方法与本研究方法的比较,(b) 本工作中用于合成碳纳米纤维的方法,(c) Ru/CNF催化剂的HAADF-STEM图像,(d) Ru/CNF催化剂和参照物的Ru K-edge XANES谱图,(e) Ru/CNF、Ru箔和RuO₂的Ru K-edge FT-EXAFS谱图。
小结:测试结果表明,Ru 高度分散在 CNF 上,无明显团聚;存在金属 Ru 与部分氧化 RuOx 共存的界面异质结构;CNF 表面羟基可诱导 Ru 发生原位自重构。
通过TEM、XANES、EXAFS三联表征,确凿证明:CNF 表面羟基驱动 Ru 形成金属 Ru / 氧化 RuOx 界面异质结构,这是后续高活性与高选择性的结构基础。
图2 | 富含木质素的玉米芯的HDO催化性能
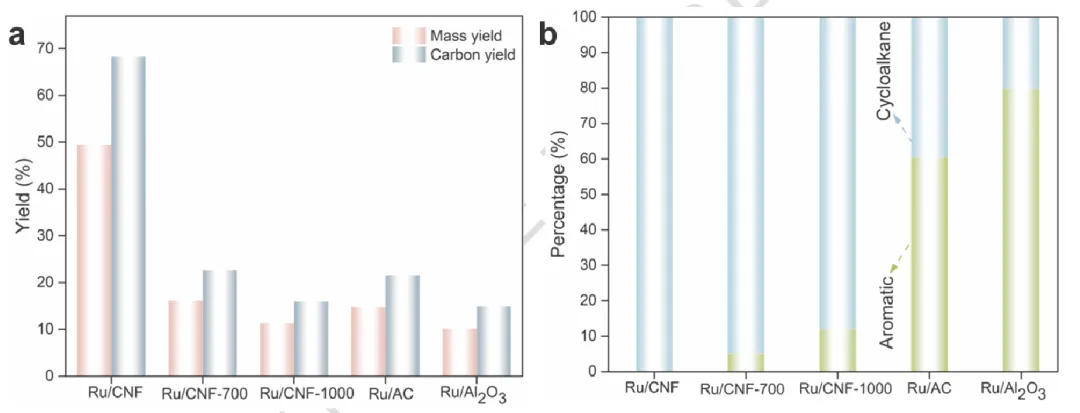
图2 富含木质素的玉米芯的HDO催化性能
(a, b) 在相同反应条件下,不同催化剂的质量收率和碳收率、产物分布的比较。反应条件:0.1g木质素,Wcat = 0.1g,20 ml十二烷,5MPa H₂,250°C,8h。
小结:Ru/CNF:质量收率 49.1%,碳收率 67.7%,产物100% 环烷烃,无芳烃。高温热处理(700/1000 ℃)脱除含氧基团后:收率大幅下降,产物出现芳烃。对比样 Ru/AC、Ru/Al₂O₃:主产物为芳烃,收率远低于 Ru/CNF
小结催化性能直接证明:Ru/RuOx 界面是定向生成环烷烃的关键;CNF 表面含氧基团决定产物选择性:富氧→环烷烃;缺氧→芳烃。
图 3 | 近环境压力X射线光电子能谱(NAP-XPS)
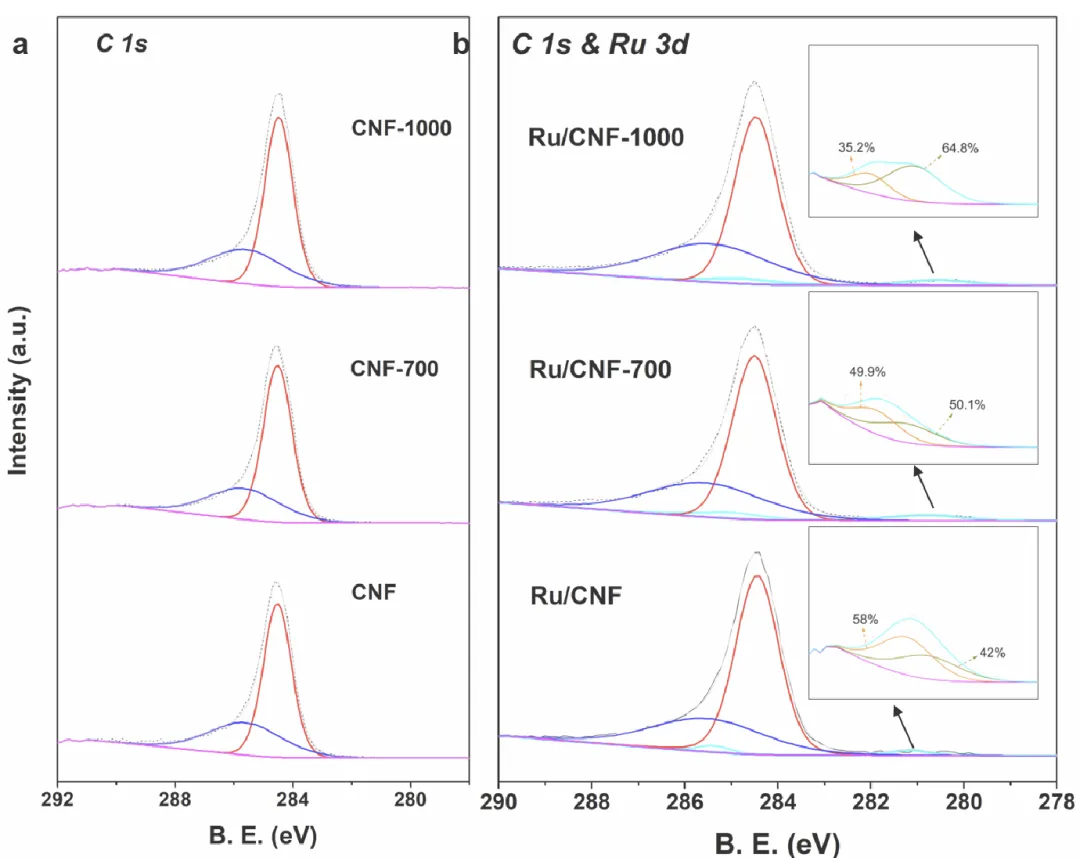
图3 近环境压力X射线光电子能谱(NAP-XPS)
(a) CNF和(b) Ru/CNF催化剂在300°C、H₂气氛(0.2 mbar)下处理1小时。
小结:Ru 3d 出现两组峰:~280.1 eV(金属 Ru)、~280.6 eV(氧化 Ru);RuOx 比例:Ru/CNF(58%) > Ru/CNF-700(49.9%) > Ru/CNF-1000(35.2%);高温脱除表面氧 → RuOx 比例下降,金属 Ru 比例上升
小结NAP–XPS 直接证实:CNF 表面含氧基团数量决定 Ru 的氧化态比例;Ru/RuOx 异质结构的相对含量可被热处理精准调控。
图 4 | Ru/CNF催化剂的分子动力学MD模拟
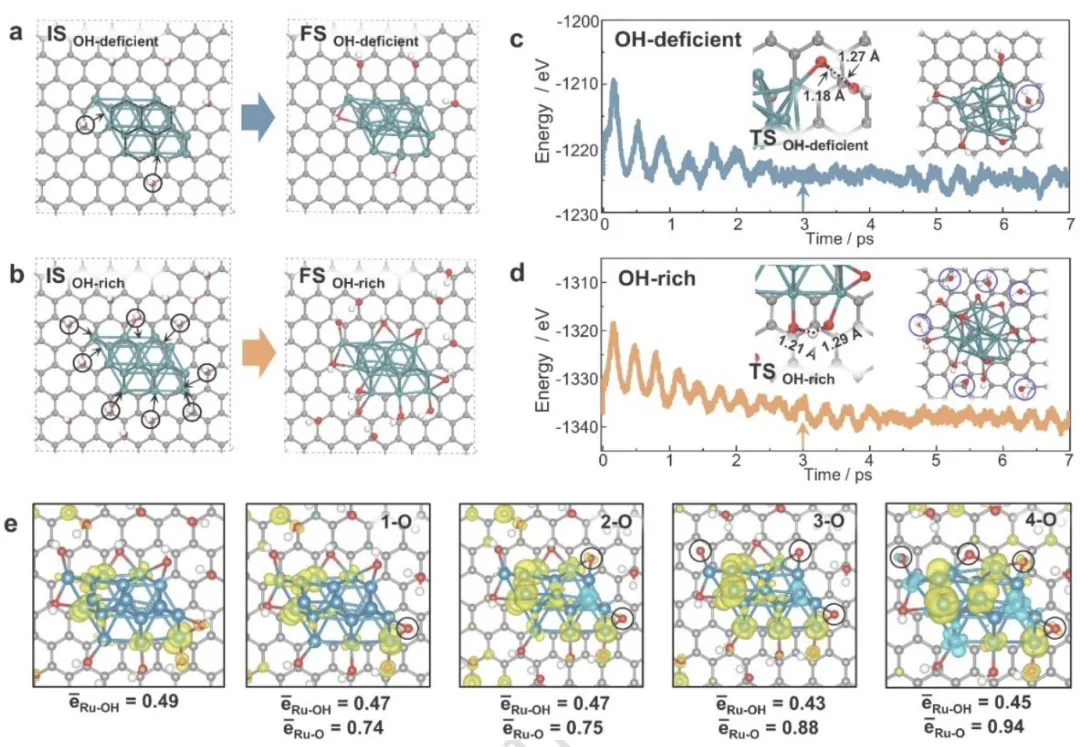
图4 Ru/CNF催化剂的分子动力学MD模拟
(a) Ru/CNF-700模型优化前后的初始态(IS)和终态(FS)结构。黑色六边形表示Ru团簇与碳纳米层之间的晶格排列。(b) Ru/CNF模型优化前后的IS和FS结构。(c) Ru/CNF-700在573 K下0 ps至7 ps的MD模拟能量分布。(d) Ru/CNF在相同时间范围和温度下的MD模拟能量分布。插图显示两个OH基团偶联形成一个H₂O和一个O原子的过渡态(TS)结构,蓝色圆圈突出显示从表面脱附的H₂O分子。箭头指示能量和温度开始围绕恒定值振荡的近似点。(e) 在Ru表面不同覆盖度下,与OH和O物种键合的Ru原子的平均Bader电荷。绿色:Ru;灰色:C;红色:O;白色:H。
小结:表面 OH 可迁移到 Ru 上,2OH → O + H₂O***,H₂O 脱附留下界面 O*;O * 强吸电子,使 Ru 形成Ruδ++・・・Ruδ+・・・Oδ−极化界面;Ru/CNF 的 Ru–O 键长更短(1.706 Å),相互作用更强;能量曲线快速稳定,表明Ru/RuOx 界面结构热力学稳定
小结MD 模拟从原子层面阐明:CNF 表面羟基在热驱动下迁移到 Ru,原位生成 Ru/RuOx 异质结,并形成强极化界面,为 H₂活化与 C–O 断裂提供位点。
图 5 | HDO中关键步骤的吉布斯自由能计算
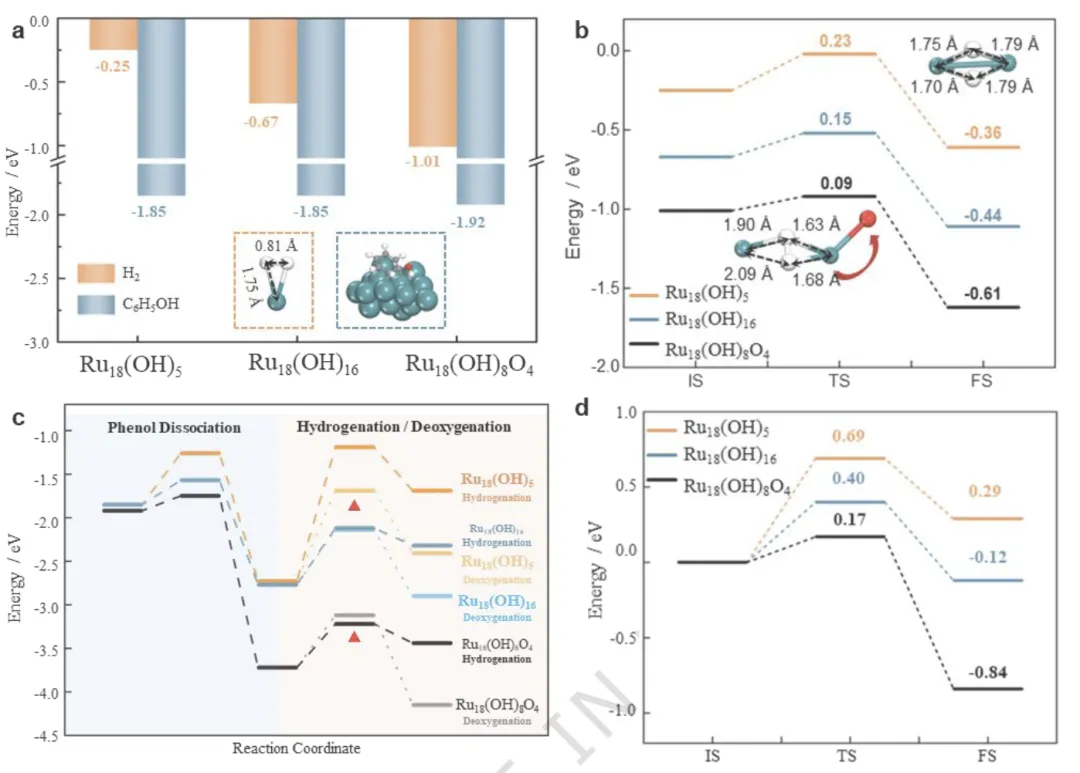
图5 HDO中关键步骤的吉布斯自由能计算
(a) H₂和苯酚在Ru₁₈(OH)₅(OH缺乏)、Ru₁₈(OH)₁₆(OH丰富)和Ru₁₈(OH)₈O₄(O修饰)模型上的吸附能。插图显示了H₂和苯酚的吸附构型,键长以黑色标示。(b) H₂解离反应的能垒分布图,插图显示过渡态结构。过渡态时的H-H键长以黑色标示。(c) 苯酚解离及随后的加氢或脱氧过程的能垒分布图。红色三角形表示优先反应路径。插图展示了脱氧反应的过渡态构型。(d) 环己醇脱氧反应的能垒分布图。插图描绘了过渡态构型。绿色:Ru;灰色:C;红色:O;白色:H。
小结:
H₂活化:富 O 表面吸附能最高(–1.01 eV),解离能垒最低(0.09 eV)
苯酚活化:富 O 表面 O–H 解离能垒最低(0.17 eV)
选择性关键:富 O 表面:苯环加氢能垒(0.50 eV)< 直接脱氧(0.60 eV)→ 优先环烷烃;
贫 OH 表面:直接脱氧能垒更低 → 优先芳烃;环己醇脱氧:富 O 表面能垒最低(0.17 eV)
小结DFT 完整揭示选择性来源:Ru/RuOx 极化界面显著降低 H₂活化、苯环加氢、脱氧三步能垒,使反应路径完全偏向环烷烃;贫氧表面则倾向直接脱氧生成芳烃。
通过热调变CNF载体表面性质以构建Ru/RuOₓ异质结构界面,所合成的Ru/CNF催化剂在木质素加氢脱氧制液态烃反应中实现了综合性能的显著提升。其活性与选择性方面,获得了高达67.7%的碳产率,并高选择性地生成饱和环烷烃,这归因于异质结构界面位点(O δ⁻•••Ru δ⁺ + •••Ru δ⁺)对H₂的异裂活化和对C-O键的强极化作用,协同降低了加氢和脱氧步骤的能垒。反应路径上,与Ru/AC等催化剂相比,Ru/CNF避免了芳香族中间体的积累,实现了更高效的直接加氢脱氧路径。稳定性方面,该催化剂在半连续操作中历经多轮循环仍保持活性不变,且反应后金属分散度和化学状态得以完好保持,展现出卓越的结构与化学稳定性。这些结果共同表明,通过理性设计金属-载体界面构建双功能活性位点,是开发高效、稳定生物质升级催化剂的有效策略。
https://doi.org/10.1038/s41467-026-71394-z
以上内容,如有误读和纰漏,敬请指正
解决世纪争议难题,郑州大学“最硬核团队”重磅Nature: 合成纯相块状六方型金刚石 院士携手!兰州大学,校史首篇Nature Nanotechnology:飞秒时间尺度上的质子 - 电子时间异步性,实现质子交换膜电解槽用耐腐蚀低铱阳极 分子筛催化,再发Nature Catalysis! 南开大学陈军院士,2025年成果汇总 最新JACS,高稳定负载型 Cu-Ni 稀释合金催化剂实现高效 CO2 重整甲烷 中科大,最新Nature Chemistry!双原子催化剂! 杰青联手!华东师范大学关小红/孙远奎&湖南大学王双印,最新Angew:硫掺杂零价铁气凝胶*NO偶联和*H供给协同调控驱动硝酸盐高效电还原成N2 湖南大学「国家杰青」王双印&中南大学任博华&天津大学杨娜,最新Angew:机器学习揭示有机-金属界面“电子海绵”行为促进CO2电还原C2+生成 李灿院士领衔!兰州大学李泽龙,最新Angew:NdO强化IrMnOx双位点协同,酸性OER低Ir长寿命! 韩布兴院士领衔!中科院化学所张裴,最新Angew!温度介导动力学与界面控制以实现浓缩热敏性生物质原料持续电氧化!

如需转载或合作,请联系我们
联系方式:15715750735(微信同)
联系邮箱:mon@xueyanhui.com
2.催化进展现有综合群、电催化交流群、同步辐射交流群、文献交流互助群、各研究领域群等近20余个,欢迎大家加小编微信,我们会尽快拉您进入对应的群。


