深静脉血栓是心血管疾病相关死亡的第三大原因,现有临床治疗(抗凝药物、血栓切除术等)虽能清除血栓,却无法有效修复内皮损伤,停药后受损内皮持续过度表达黏附分子,介导病理性血小板-内皮相互作用,导致约15%的复发率。因此,如何同时阻断病理性血小板黏附并修复内皮功能,是改善深静脉血栓预后的核心难题。
南京大学鼓楼医院李晓强团队通过单细胞测序首次揭示cGAS-STING-NLRP3轴是驱动深静脉血栓内皮损伤和病理性血小板黏附的关键通路,并基于人工智能辅助药物筛选鉴定连翘酯苷A为新型STING抑制剂。在此基础上,团队创新性构建了负载连翘酯苷A的血小板膜仿生纳米凝胶Pm@Fng,该人造血小板通过GPIbα-vWF介导的竞争性黏附实现血栓部位主动靶向,在酸性与高活性氧微环境中响应性释放药物,从物理阻断和化学调控双重维度抑制血栓形成并修复内皮功能。相关成果以“Artificial platelets suppressing deep vein thrombosis via competitive adhesion”发表在《Bioactive Materials》期刊上。
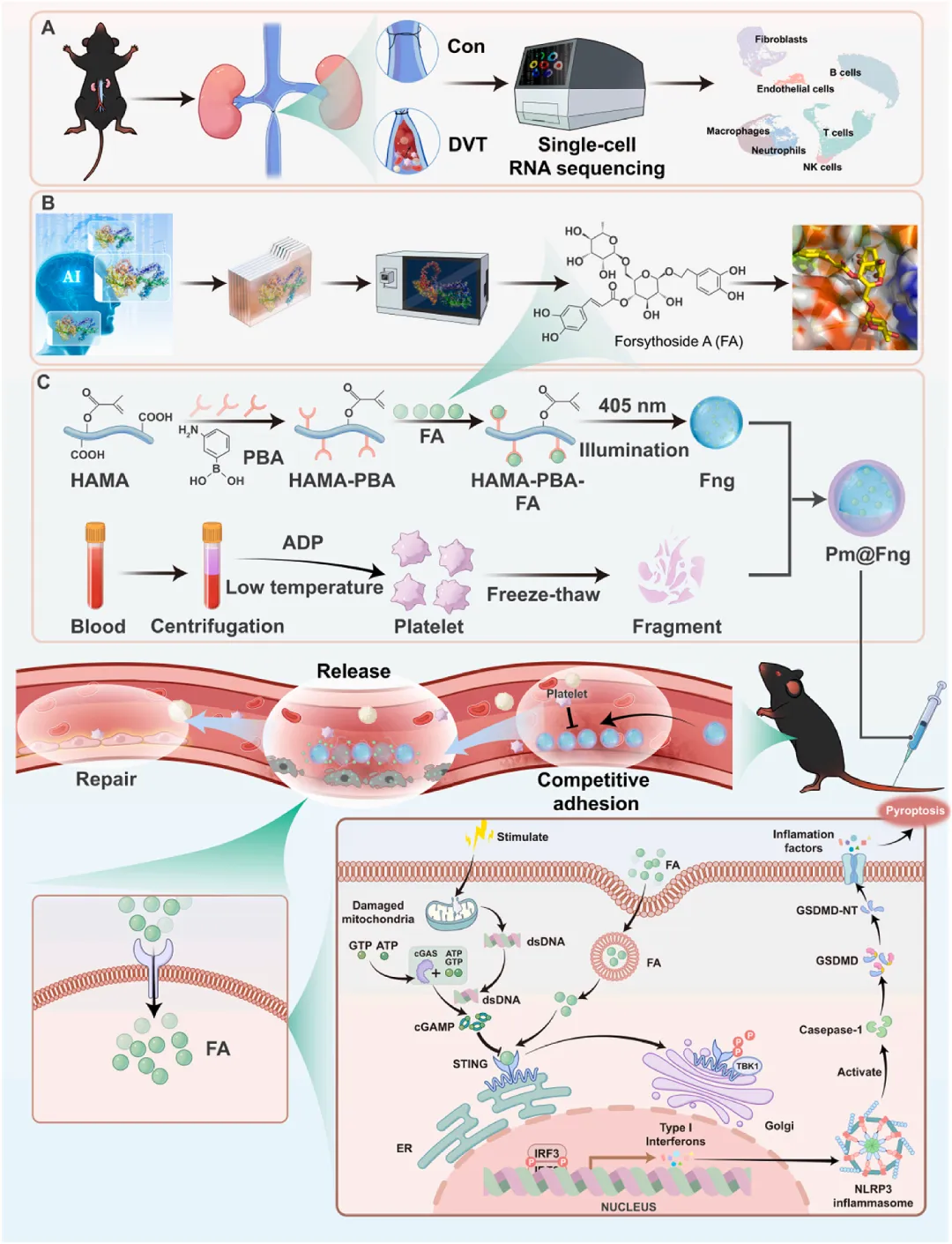
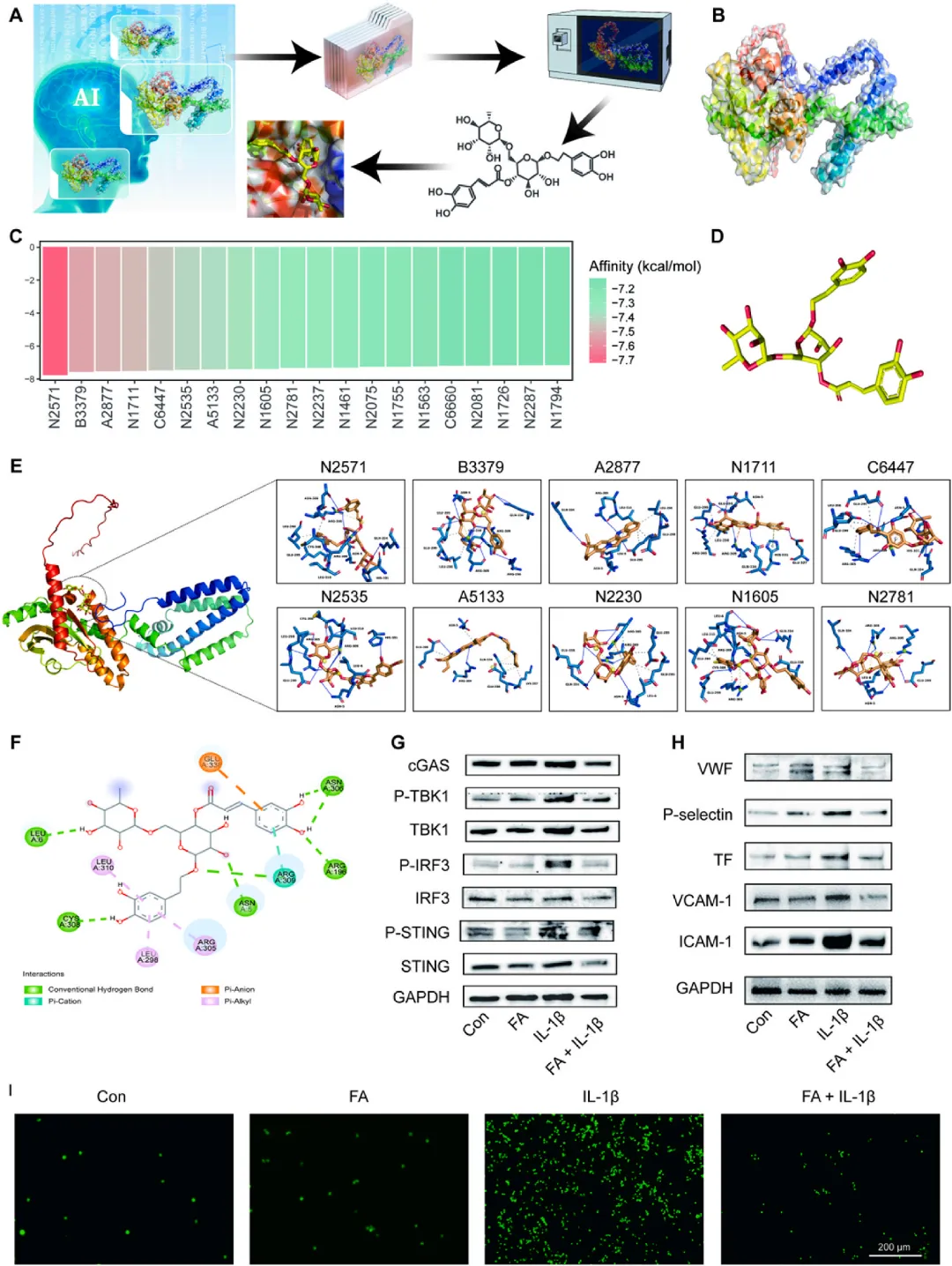
示意图1 单细胞测序揭示深静脉血栓机制、人工智能辅助筛选STING抑制剂及人造血小板Pm@Fng的构建与作用机制
该研究首先对小鼠下腔静脉狭窄模型的血栓组织进行单细胞RNA测序,解析深静脉血栓微环境中的分子调控网络。随后,利用AlphaFold2预测STING蛋白活性构象,结合深度学习驱动的分子对接平台,从807种临床药物中筛选出连翘酯苷A作为STING抑制剂。最后,通过光聚合-膜挤出技术构建人造血小板Pm@Fng:将连翘酯苷A负载于透明质酸-苯硼酸纳米凝胶核心,外包覆活化血小板膜囊泡。经静脉注射后,Pm@Fng通过GPIbα-vWF轴靶向损伤内皮,竞争性占据黏附位点以阻断天然血小板募集;在酸性和高活性氧的血栓微环境中,苯硼酸酯键断裂释放连翘酯苷A,抑制cGAS-STING-NLRP3信号通路,减轻血管功能障碍并有效抑制血栓形成。
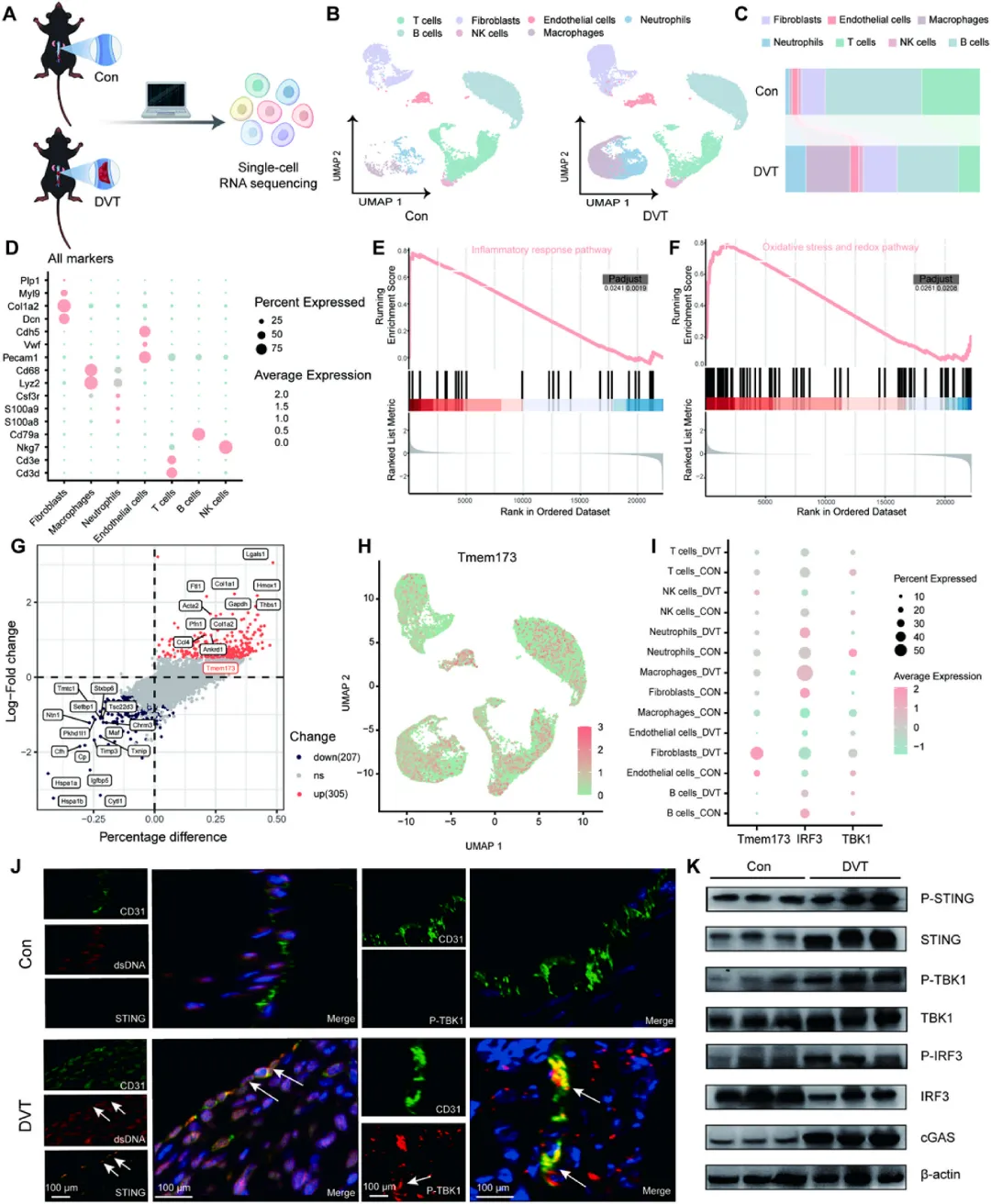
图1 单细胞图谱揭示深静脉血栓形成过程中的分子调控网络
对下腔静脉狭窄术后7天的小鼠血栓组织进行单细胞RNA测序,识别出成纤维细胞、内皮细胞、T细胞、B细胞、NK细胞、中性粒细胞和巨噬细胞等七种主要细胞群体。与假手术对照组相比,深静脉血栓组中性粒细胞比例从2.07%升至10.65%,巨噬细胞从1.46%升至22.80%,而B细胞和T细胞比例下降,表明早期血栓微环境以先天免疫细胞快速浸润为特征。基因集富集分析显示炎症反应与氧化应激通路显著上调,差异表达基因分析发现TMEM173(编码STING蛋白)在深静脉血栓组显著高表达,且特异性富集于内皮细胞亚群。免疫荧光共定位和蛋白印迹实验证实,深静脉血栓组内皮细胞中双链DNA损伤信号、STING及其磷酸化形式、TBK1和IRF3均显著激活。术后第2天的补充分析进一步确认STING通路在血栓形成早期即已活化,表明其是深静脉血栓发病的早期驱动因素,而非仅为慢性血栓的特征性表现。
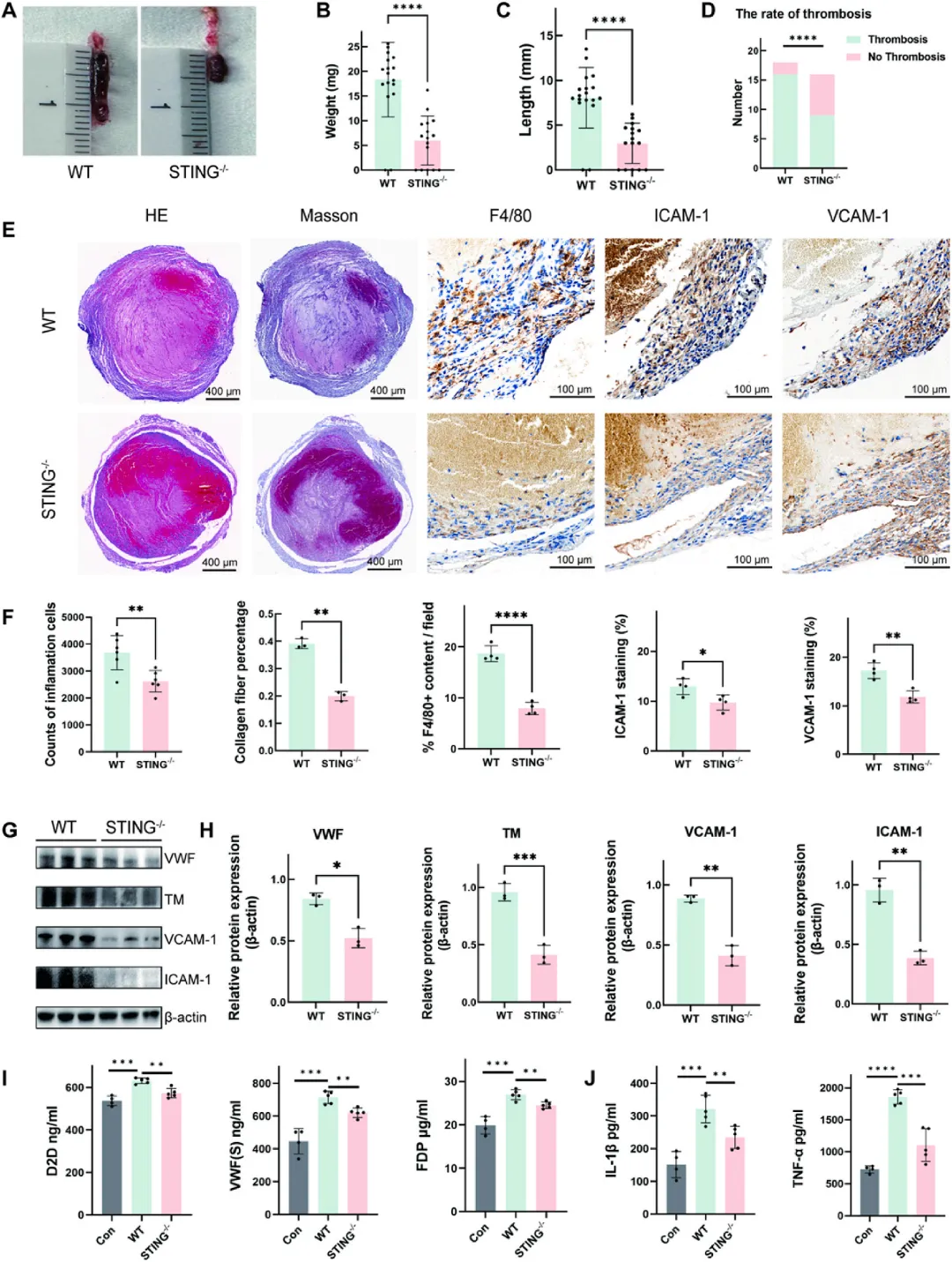
图2 STING基因缺失抑制深静脉血栓形成
STING敲除小鼠在下腔静脉狭窄术后7天,血栓体积、重量(从20.63 mg降至8.69 mg)和长度(从9.08 mm降至3.96 mm)均显著减少,血栓发生率从88.89%降至56.25%。组织学分析显示敲除小鼠血栓内F4/80阳性巨噬细胞浸润减少,ICAM-1和VCAM-1表达显著下调,血浆中vWF、D-二聚体和FDP水平降低,而抗凝蛋白TM相对上调。此外,STING缺失还抑制了NLRP3炎症小体活化及下游焦亡相关蛋白表达。上述结果表明STING通过调控内皮黏附分子表达、炎症细胞募集、凝血-抗凝平衡及内皮焦亡等多重机制促进深静脉血栓形成,其基因缺失可从多维度抑制血栓发生发展。
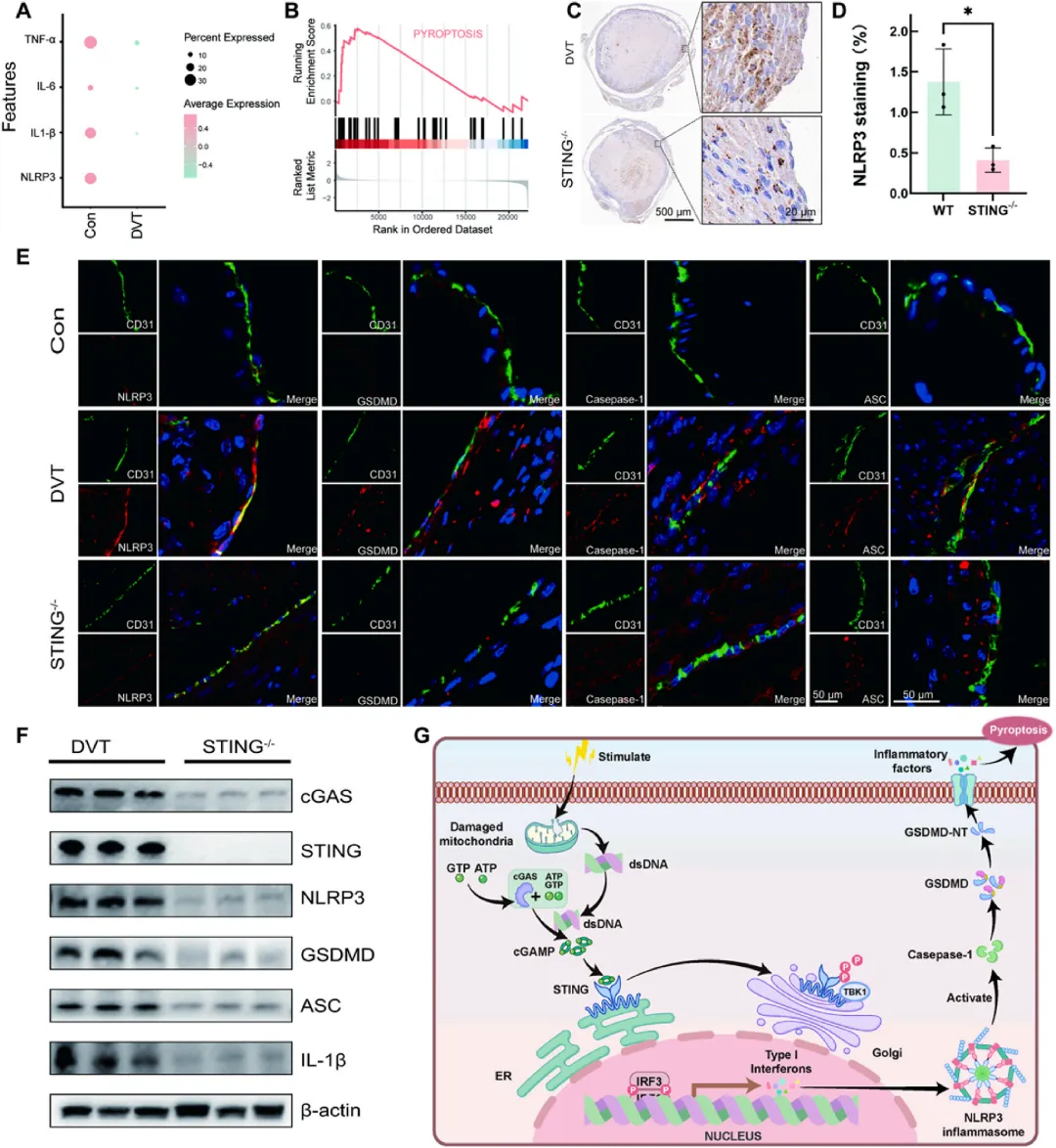
图3 STING缺陷抑制NLRP3炎症小体激活与内皮细胞焦亡
单细胞测序显示深静脉血栓组内皮细胞中NLRP3、IL-1β和TNF-α表达显著上调,焦亡信号通路富集明显。免疫荧光共定位证实NLRP3、GSDMD、Caspase-1和ASC与内皮标志物CD31共定位,而STING敲除后这些焦亡相关蛋白表达显著下降。蛋白印迹进一步验证STING缺失可抑制NLRP3、ASC、GSDMD和IL-1β的表达。体外实验中,IL-1β刺激C166内皮细胞可诱导STING磷酸化及NLRP3表达上调,使用STING抑制剂H-151可阻断TBK1磷酸化及下游焦亡标志物,而NLRP3抑制剂MCC950仅阻断GSDMD-N形成和IL-1β剪切而不影响上游P-TBK1水平,证实STING信号位于NLRP3炎症小体上游。机制上,内皮细胞损伤释放线粒体DNA激活cGAS-STING轴,进而通过TBK1-IRF3信号促进NLRP3炎症小体活化,诱导GSDMD介导的焦亡和炎症因子风暴,破坏内皮屏障功能并释放更多促血栓因子,间接加剧血小板黏附与血栓稳定。
图4 STING特异性抑制剂的筛选与验证
利用AlphaFold2预测小鼠STING蛋白活性构象,构建动态药效团模型识别cGAMP结合域和二聚化界面等关键互作位点。采用深度神经网络驱动的分子对接平台,基于结合自由能(≤-7.0 kcal/mol)筛选807种临床药物,获得20种候选分子,其中连翘酯苷A因最佳结合亲和力(-7.718 kcal/mol)被鉴定为最优STING抑制剂。结构分析显示连翘酯苷A通过氢键、π-阴离子和π-烷基等多重非共价相互作用与STING蛋白结合。表面等离子体共振实验证实连翘酯苷A与STING蛋白直接结合,解离常数为18.2 μM。体外实验中,连翘酯苷A(40 μM)显著抑制IL-1β诱导的STING、TBK1和IRF3磷酸化,并下调vWF、P-选择素、组织因子、VCAM-1和ICAM-1等黏附与促凝分子表达,同时减少巨噬细胞向内皮单层的黏附。cGAMP刺激和STING靶向siRNA实验进一步证实连翘酯苷A的抗炎作用依赖于STING通路抑制。
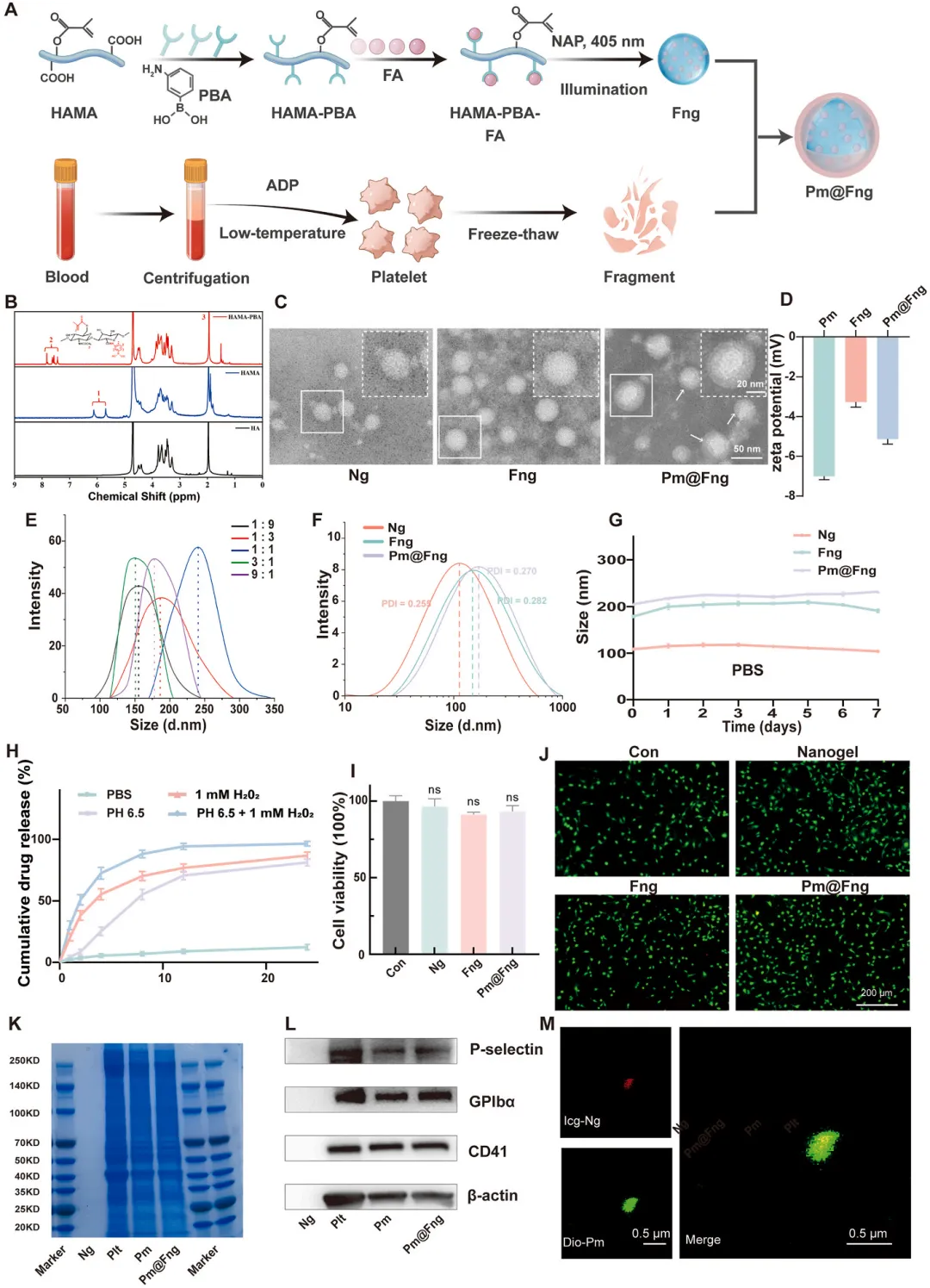
图5 人造血小板Pm@Fng的合成与表征
通过两步化学修饰透明质酸(甲基丙烯酸酯化和苯硼酸化)构建功能化聚合物骨架,核磁共振氢谱证实双官能团化成功。连翘酯苷A与苯硼酸基团在温和条件下形成动态硼酸酯键,经光引发自由基聚合制备三维交联纳米凝胶,最后通过静电吸附与光交联策略包覆活化血小板膜囊泡。透射电镜显示Pm@Fng呈核壳结构,纳米凝胶核心约50 nm,膜壳厚度约8 nm,水合粒径约205 nm,Zeta电位介于裸纳米凝胶与纯膜囊泡之间,证实膜包覆成功。高效液相色谱表明药物包封率为73.5%,载药量为19.7%。在pH 7.4生理环境中24小时累积释放率仅12.3%,而在pH 6.5和1 mM H₂O₂条件下释放显著增加,双刺激同时作用下4小时释放率达72.5%、24小时达96.5%,展现出优异的血栓微环境响应性释放特性。CCK-8和活死染色证实材料无细胞毒性,蛋白电泳和免疫印迹确认血小板膜表面功能蛋白(P-选择素、CD41、GPIbα)完整保留。
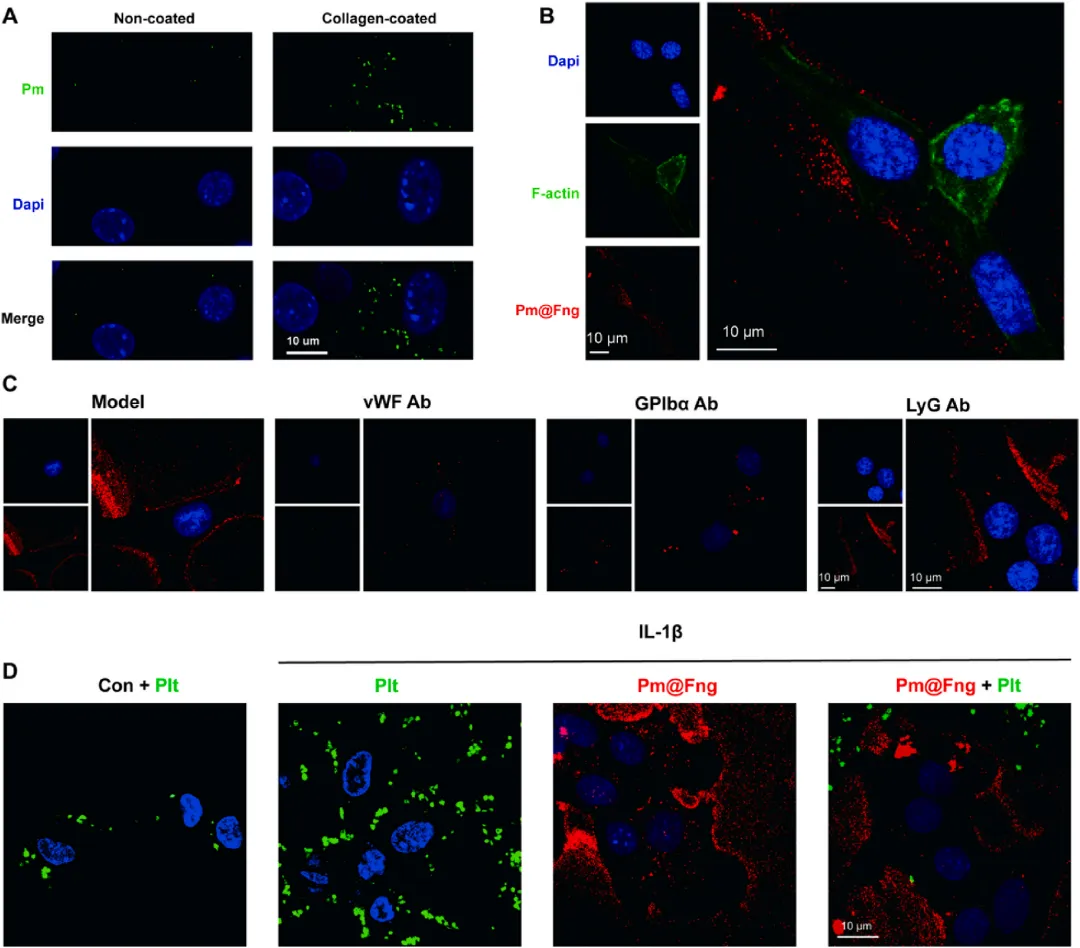
图6 Pm@Fng靶向能力与竞争性黏附机制的体外验证
体外结合实验显示,ICG标记的Pm@Fng可特异性结合vWF包被表面和I型胶原,该结合可被抗GPIbα抗体显著阻断,证实其依赖GPIbα-vWF轴。在IL-1β活化内皮细胞黏附实验中,抗vWF或抗GPIbα抗体预处理均可显著减少Pm@Fng的结合。竞争性黏附实验中,Pm@Fng与钙黄绿素标记的天然血小板共孵育后,可显著抑制天然血小板向活化内皮细胞的黏附,直接证明人造血小板通过竞争性占据vWF结合位点阻断病理性血小板募集。
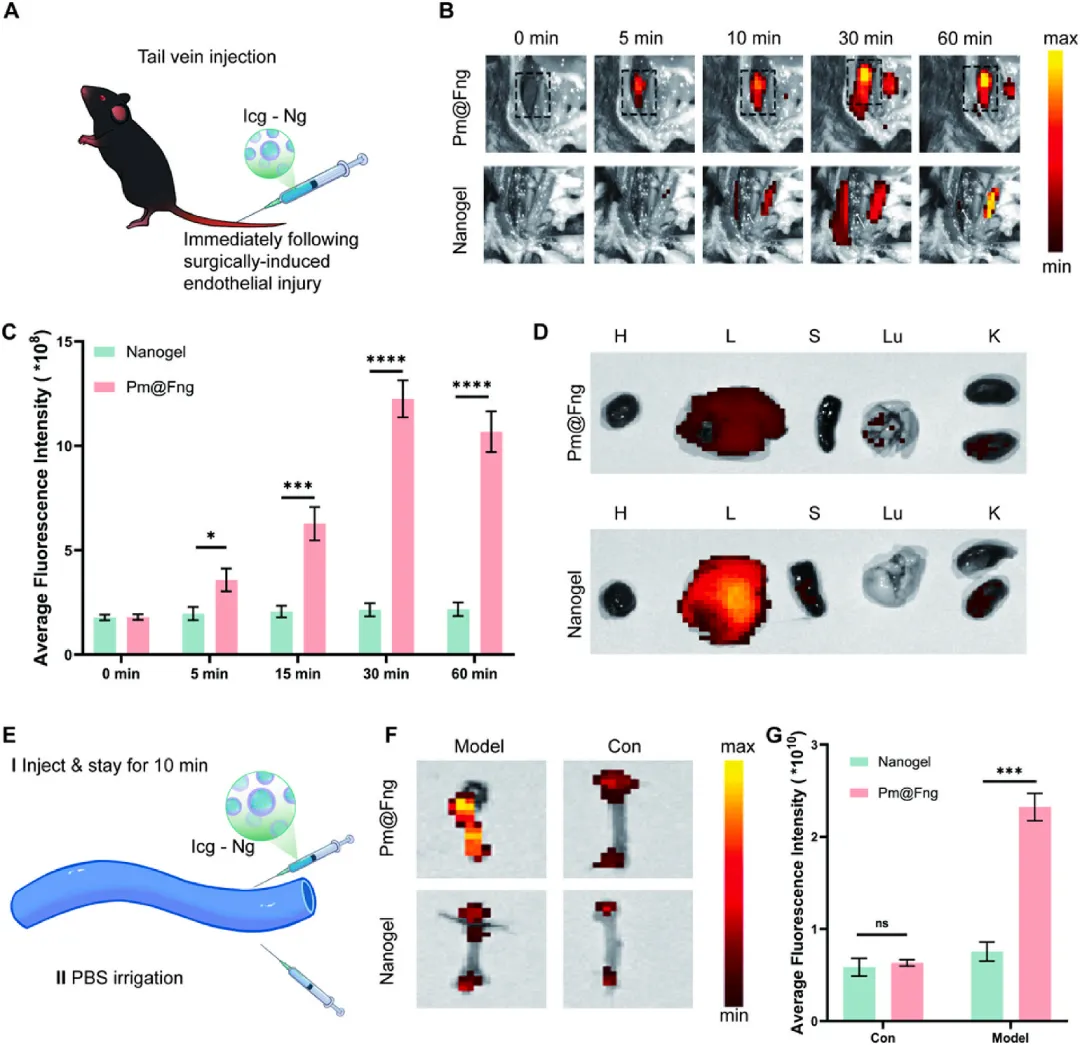
图7 Pm@Fng在体内优先靶向损伤内皮并富集于血栓部位
在小鼠下腔静脉狭窄模型中,静脉注射ICG标记的Pm@Fng后5分钟即在血栓部位出现强荧光信号,30分钟达峰值,荧光强度为裸纳米凝胶组的4倍,随后信号逐渐下降。离体器官成像显示24小时后肝脏和肾脏为主要代谢器官,脾脏蓄积较低,提示血小板膜表面CD47等蛋白帮助逃避脾脏巨噬细胞吞噬清除,延长循环时间。体外血管段孵育实验进一步证实Pm@Fng在损伤血管段显著富集,而裸纳米凝胶结合微弱,表明人造血小板可通过GPIbα-vWF特异性结合和血流动力学介导的被动滞留双重机制实现血栓部位精准靶向。
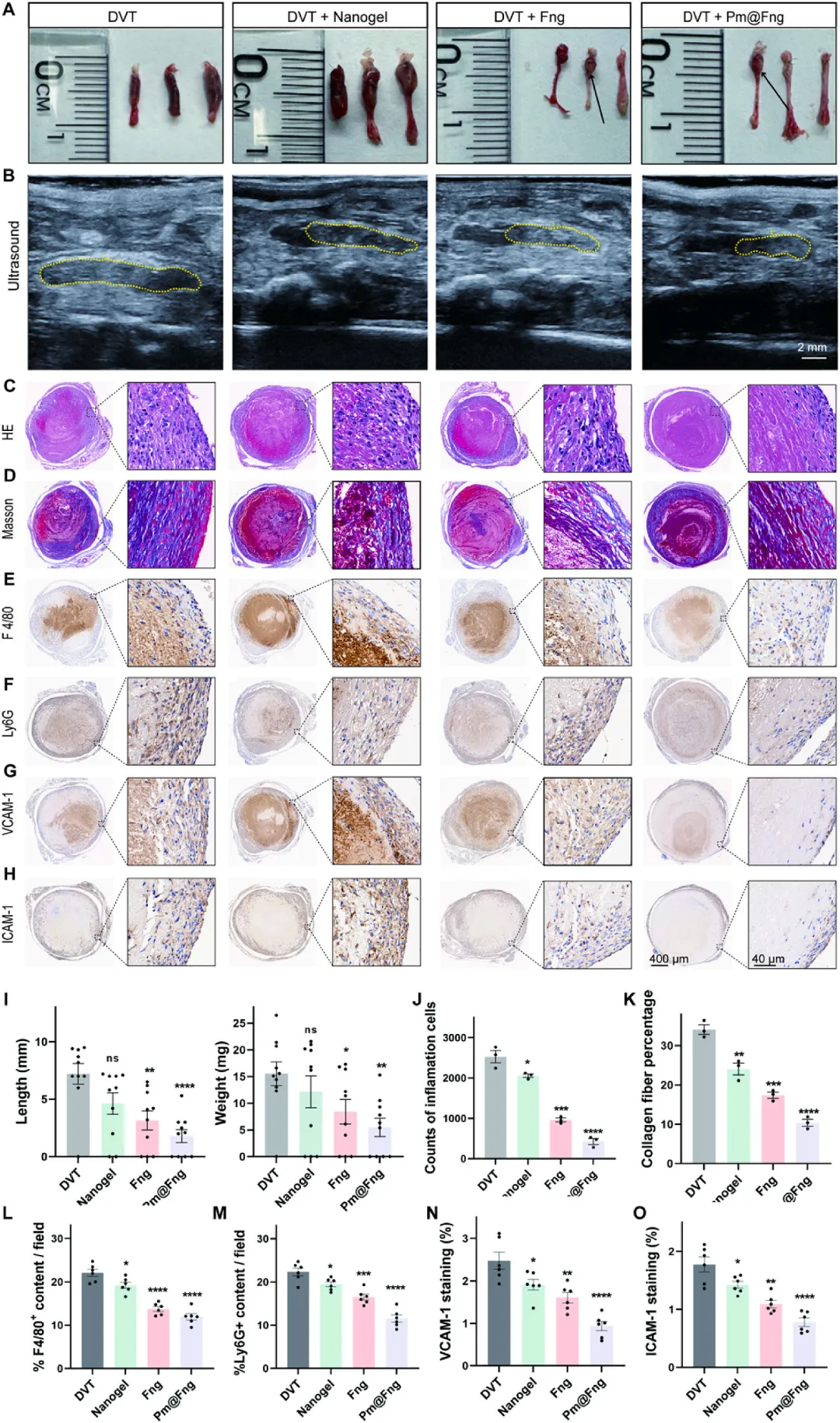
图8 Pm@Fng有效抑制深静脉血栓形成
在下腔静脉狭窄术后连续7天每日尾静脉给药方案中,Pm@Fng组血栓发生率从90%降至60%,血栓重量从17.29 mg降至9.10 mg,长度从8.01 mm降至2.98 mm。超声影像证实Pm@Fng组血栓体积最小。苏木精-伊红染色显示Pm@Fng组血管壁结构完整,内皮连续性保持良好,平滑肌层水肿和炎症细胞浸润显著减轻;Masson染色显示Pm@Fng组血栓内纤维蛋白沉积面积明显减少。免疫组化分析表明Pm@Fng组F4/80阳性巨噬细胞和Ly6G阳性中性粒细胞浸润显著抑制,VCAM-1和ICAM-1表达下调,证实人造血小板通过竞争性黏附阻断病理血小板-内皮相互作用,同时减轻局部炎症反应,实现高效抗血栓效果。
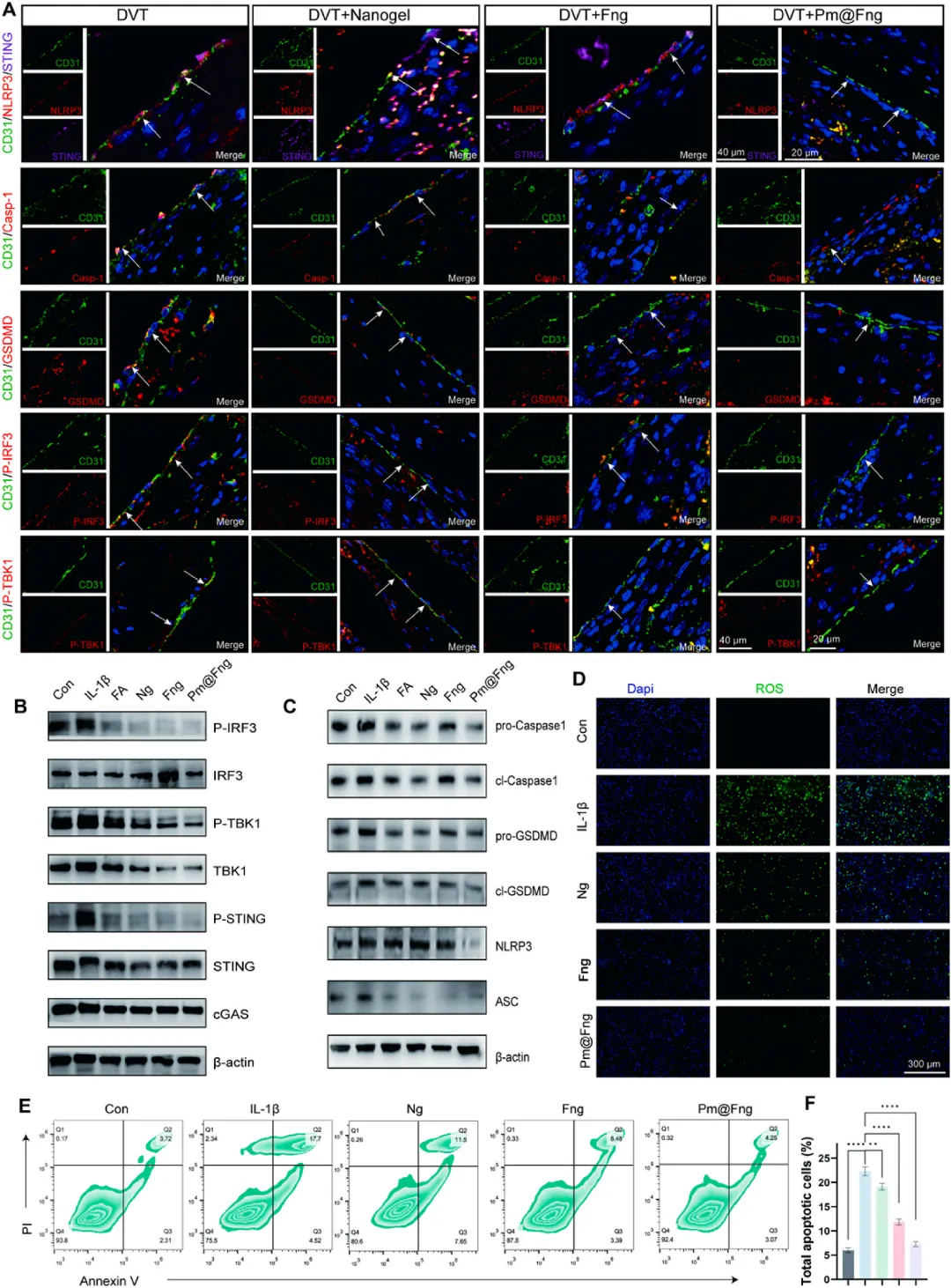
图9 Pm@Fng通过cGAS-STING-NLRP3串扰恢复内皮稳态
免疫荧光染色显示Pm@Fng处理组深静脉血栓内皮细胞中NLRP3、STING、cleaved Caspase-1、GSDMD、P-TBK1和P-IRF3表达显著降低。蛋白印迹实验进一步证实Pm@Fng可抑制IL-1β诱导的cGAS-STING-TBK1-IRF3轴激活,降低NLRP3、ASC、cleaved Caspase-1和GSDMD等焦亡标志物水平。DCFH-DA活性氧探针显示Pm@Fng显著降低IL-1β刺激后内皮细胞内活性氧水平,流式细胞术Annexin V/PI双染证实其减少内皮细胞凋亡。综合结果表明,人造血小板通过连翘酯苷A靶向释放抑制STING-NLRP3焦亡通路,同时利用苯硼酸酯的活性氧响应特性清除氧化应激,实现内皮功能的多维度修复。
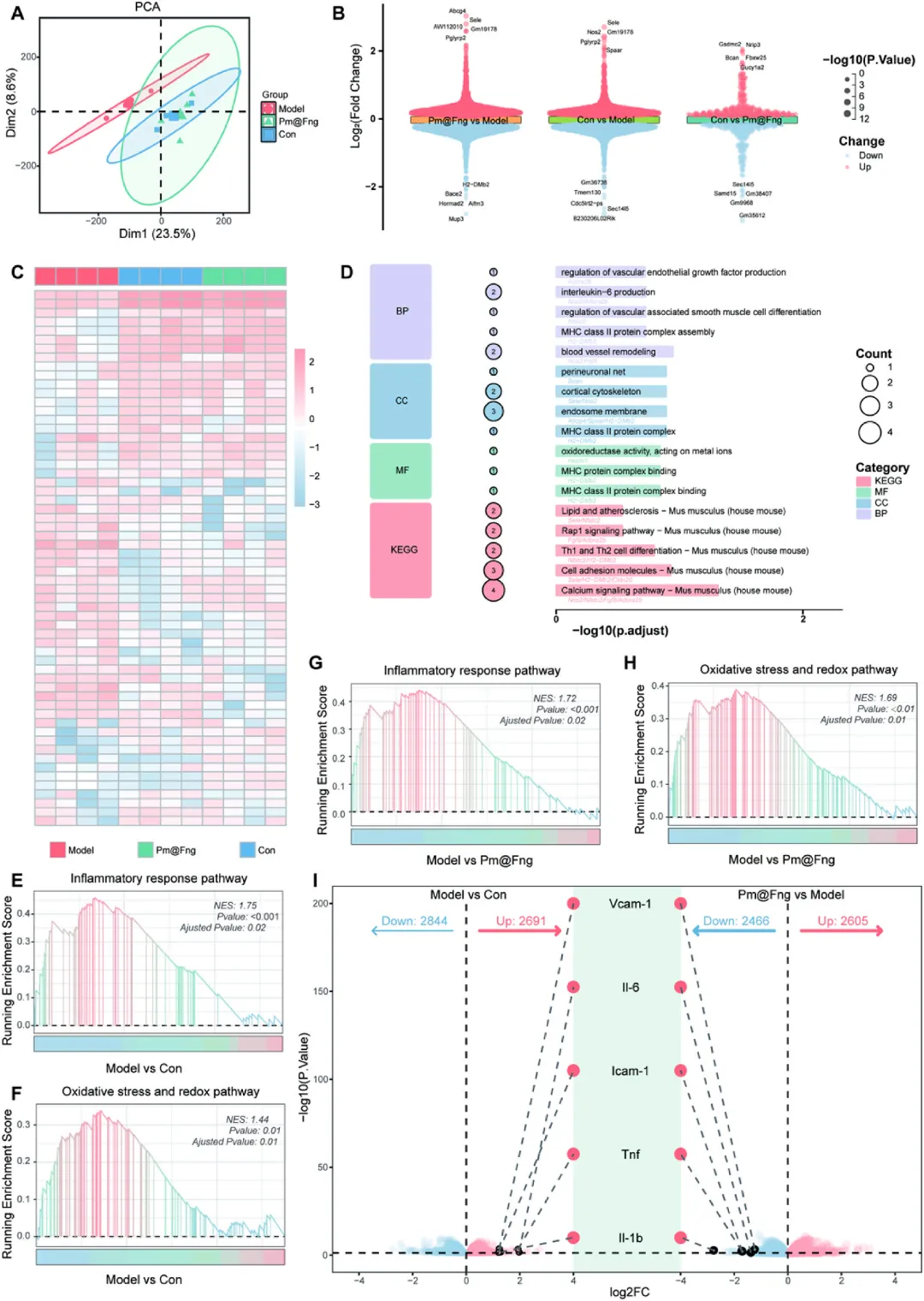
图10 Pm@Fng介导内皮表型重编程的转录组学证据
对正常对照组、IL-1β诱导血栓微环境模型组和Pm@Fng处理组内皮细胞进行RNA测序,主成分分析与层次聚类显示Pm@Fng组转录谱显著向正常对照组趋近,与模型组明显分离。GO和KEGG富集分析表明,Pm@Fng逆转了炎症反应、氧化应激和细胞黏附相关通路的异常激活,同时上调PI3K-Akt信号通路和血管重塑通路。差异基因分析显示,模型组中上调的IL-1β、TNF、IL-6、ICAM-1和VCAM-1在Pm@Fng组被显著抑制。上述结果证实人造血小板通过竞争性黏附介导的靶向干预,可将内皮细胞从“促炎/促黏附”表型重编程为“抗炎/抑黏附/促修复”表型,从转录水平验证了其治疗价值。
综上,本研究首次系统阐明了cGAS-STING-NLRP3轴在深静脉血栓内皮损伤中的核心驱动作用,并通过人工智能辅助筛选鉴定连翘酯苷A为新型STING抑制剂。在此基础上构建的仿生人造血小板Pm@Fng,将GPIbα-vWF介导的竞争性黏附物理阻断与STING-NLRP3通路化学调控有机结合,在深静脉血栓小鼠模型中将血栓形成率从90%降至60%,显著减少血栓负荷并维护血管完整性。该策略突破了传统抗凝治疗“只抗凝不修复”的局限,从物理和信号双重维度重建血管微环境,为深静脉血栓及更广泛的内皮损伤相关疾病提供了全新的治疗范式,具有重要的临床转化前景。