Single-Atom Ni on Tungsten Oxides Drives Hydrogen Spillover for Efficient Plastic Waste Upcycling
负载于氧化钨上的单原子镍驱动氢溢流以实现塑料废弃物高效升级回收https://doi.org/10.1021/acscatal.5c06543通过加氢裂化将聚烯烃废弃物升级转化为液体燃料,需要兼具强酸性和加氢活性且经济高效的催化剂,这就要求对催化位点进行精确的原子级调控。本研究报道了一种非贵金属催化剂,该催化剂由原子级分散的镍负载于结构工程化处理的WO₂.₇₂纳米线上构成,其中异裂型Ni-O-W位点可实现氢气的异裂活化及氢溢流。原位红外光谱表明,在氢溢流过程中,末端W═O物种可动态转化为具有活性的布朗斯台德酸性W-OH基团,相较于WO₃中的桥连W-OH-W物种,该基团更有利于C-C键的断裂。优化后的1Ni/WO₂.₇₂催化剂在240℃下可实现聚乙烯的完全转化,对汽油和航空燃料范围的液体燃料的选择性达94.3%,液体燃料产率高达5.0 g液体燃料/(g催化剂·h),性能优于贵金属催化剂。时分辨原位红外光谱揭示了加氢裂化反应的逐步路径,包括脱氢、质子化和C-C键断裂;动力学研究和密度泛函理论(DFT)建模证实了这些工程化位点的关键作用。本研究通过氧化物结构的原子级工程化处理及金属核数调控,建立了单原子非贵金属双功能催化剂的设计策略,为塑料废弃物升级转化提供了机理洞察和实践指导。城市塑料废弃物中超过60%为聚烯烃,其全球年产量超过1亿吨。聚烯烃的化学惰性和抗降解性引发了严重的环境问题。将聚烯烃催化转化为高附加值燃料或化工原料,提供了一种可持续且经济可行的解决方案。
加氢裂化可将长链烃分解为轻质烷烃,已成为塑料升级转化的有前景的途径。该过程的关键在于开发双功能催化剂,其兼具金属位点(用于(脱)加氢)和强酸性位点(以实现C-C键断裂)。氧化钨和硫化钨作为强酸性组分,被广泛用作加氢裂化和加氢异构化反应的助催化剂,并已拓展至塑料转化领域。例如,Pt/WOx/ZrO₂ + HY催化剂可在250℃下从聚烯烃获得>85%的液体产率, 而我们前期采用Ni-WO₃/Al₂O₃ + Beta催化剂在280℃下实现了86.2%的C₅-₁₆燃料产率。 此外,基于WOx的催化剂(尤其是与Pt或Pd等贵金属复合时)通过氢溢流增强布朗斯台德酸性,可实现香草醛和甘油等生物基平台分子的加氢转化。在这些体系中,WOx发挥双重作用:W⁶⁺中心作为路易斯酸位点,而在富氢条件下,布朗斯台德酸位点通过金属位点的氢溢流形成。 然而,这些催化WOx物种通常通过简单的湿浸渍法负载于载体上,缺乏对其结构和电子性质的精确调控,因而往往依赖贵金属或强酸性沸石组分。
氢溢流——即解离的氢原子从金属位点迁移至载体——是H₂气氛下WOx表面原位生成布朗斯台德酸的关键机制。WOx接受并活化氢物种的能力取决于其形貌、氧化态和缺陷化学。例如,二维纳米片Pt/WO₃催化剂可在250℃下高效将低密度聚乙烯转化为C₅-₂₀液态烷烃,其高活性归因于纳米片结构带来的丰富表面缺陷和增强的氢溢流。更广泛地,WOx基体系中布朗斯台德酸的形成源于两个协同因素:(i) 金属位点高效活化氢;(ii) 富含缺陷的WOx结构域将氢稳定为表面−OH物种。对Pt/γ-WO₃的密度泛函理论(DFT)研究进一步支持了这一机制,表明Pt上自发解离的H₂随后迁移至氧缺陷WO₃结构域,在高温下生成表面质子物种和氧空位。除这些开创性研究外,氢溢流过程可能生成不同类型的羟基(如桥连或末端羟基),其酸性受W-OH键结构和极性的显著影响。尽管WOx的结构设计对其酸性有重要影响,但活性WOx结构域的精准识别及其在原位氢溢流条件下的演变机制仍不明确。
此外,以非贵金属Ni替代常用的Pt或Pd虽具有显著成本优势,但因Ni的氢活化能力较低且在反应条件下易团聚,面临重大挑战。这些局限性限制了可迁移氢物种的形成,进而抑制了Ni-WOx体系中的布朗斯台德酸性。为解决这一问题,构建原子级分散的Ni-WOx界面可形成Ni-O-W配位中心,促进高效的异裂氢活化与溢流,同时通过强金属-载体相互作用稳定Ni原子。然而,此类体系在聚烯烃加氢裂化及传统石油基原料转化中的应用仍鲜有探索,尤其是关于局部Ni-WOx结构(包括形貌、氧化态和缺陷性质)如何协同调控氢溢流、酸位点形成及原子级非贵金属位点的作用机制。
本研究报道了一种1Ni/WO₂.₇₂催化剂,其具有精确调控的纳米线结构、价态和单原子Ni分散性。该催化剂在240–260℃下可实现聚乙烯完全转化为C₅-₂₀烷烃,选择性达94.3%,燃料生产速率为5.0 g液体燃料/(g催化剂·h)。其液体燃料产率优于贵金属和沸石基体系,归因于其无孔结构和优化的金属氧化物界面。深入的谱学和理论研究揭示,高活性布朗斯台德W-OH位点源于末端W═O基团通过原子级分散Ni-O-W位点的氢溢流转化而来,而传统1Ni/WO₃中的Ni纳米颗粒和桥连W-OH-W需更高温度才能达到相当活性。这些发现强调了精确调控金属氧化物界面原子结构对释放非贵金属催化剂潜力的关键作用。本研究提出了一种普适且经济的策略,用于设计高性能催化剂以实现可持续塑料升级转化。
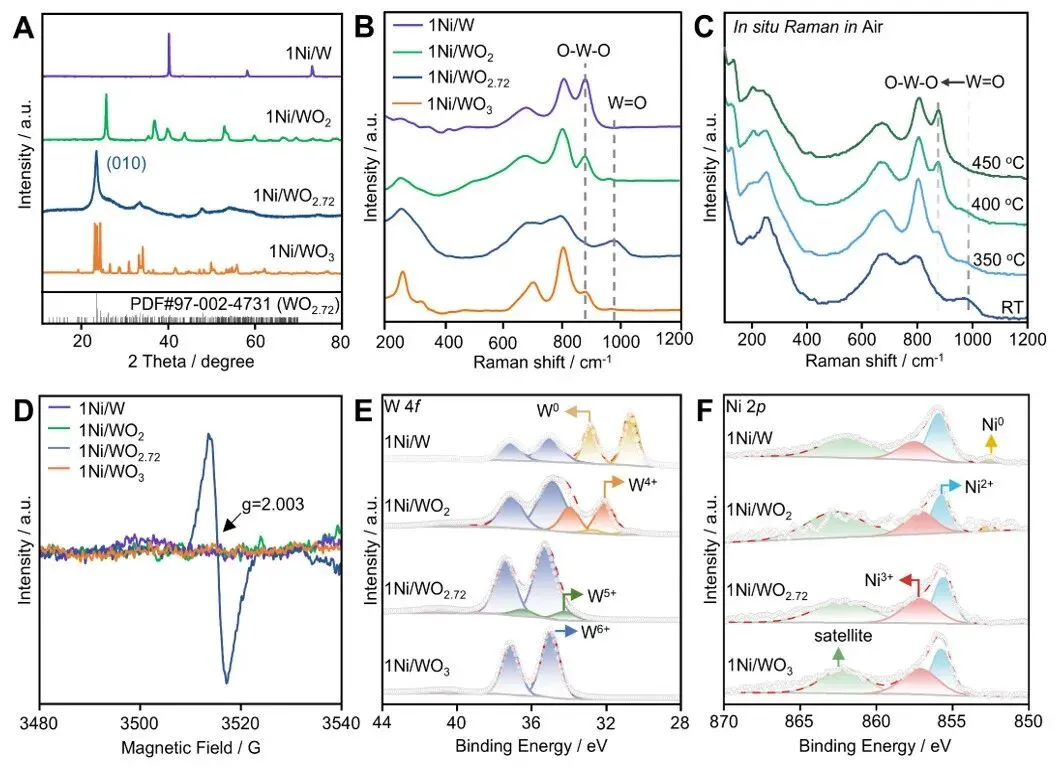
图1. 催化剂表征。(A)X射线衍射(XRD)图谱;(B)1Ni/WOx(x = 0, 2, 2.72, 3)的拉曼光谱;(C)空气中加热至450℃时1Ni/WO₂.₇₂的原位拉曼光谱;(D)电子顺磁共振(EPR)光谱及(E)W 4f和(F)Ni 2p的X射线光电子能谱(XPS)。
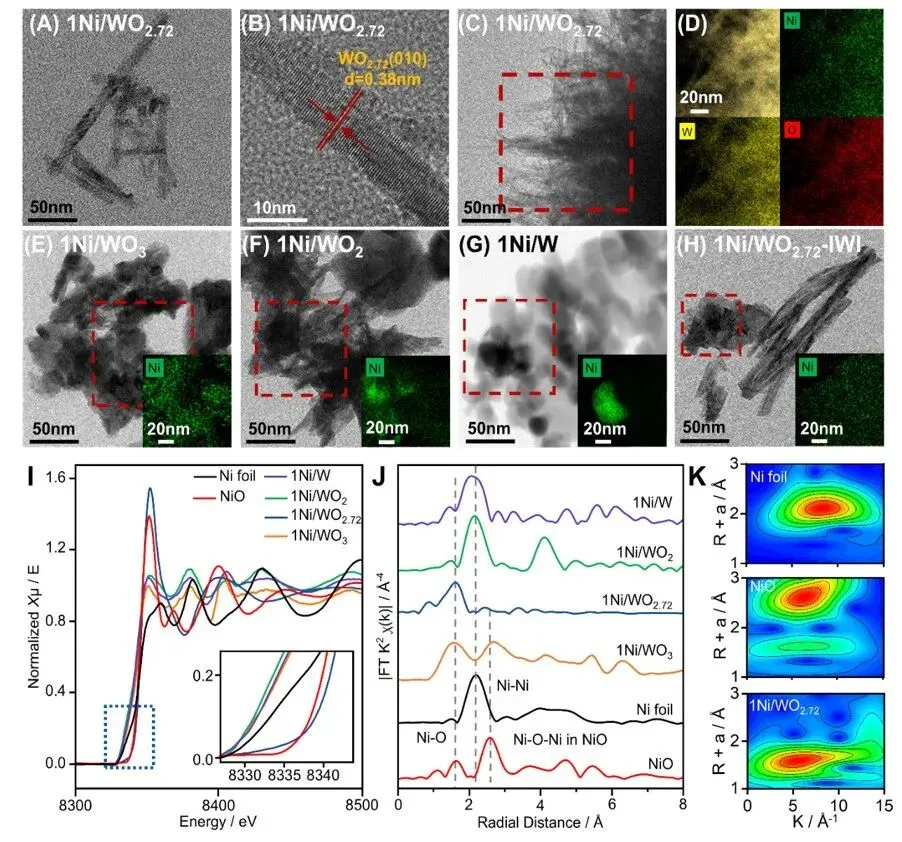
图2. 镍物种表征。(A–H)1Ni/WOx(x = 0, 2, 2.72, 3)及1Ni/WO₂.₇₂-IWI的高角环形暗场扫描透射电子显微镜(HAADF-STEM)与能量色散X射线(EDX)映射图像;(I)Ni箔、NiO及1Ni/WO₂.₇₂的Ni K边X射线吸收近边结构(XANES)光谱;(J)R空间扩展X射线吸收精细结构(EXAFS)光谱;(K)k³加权EXAFS信号的小波变换(WT)图。
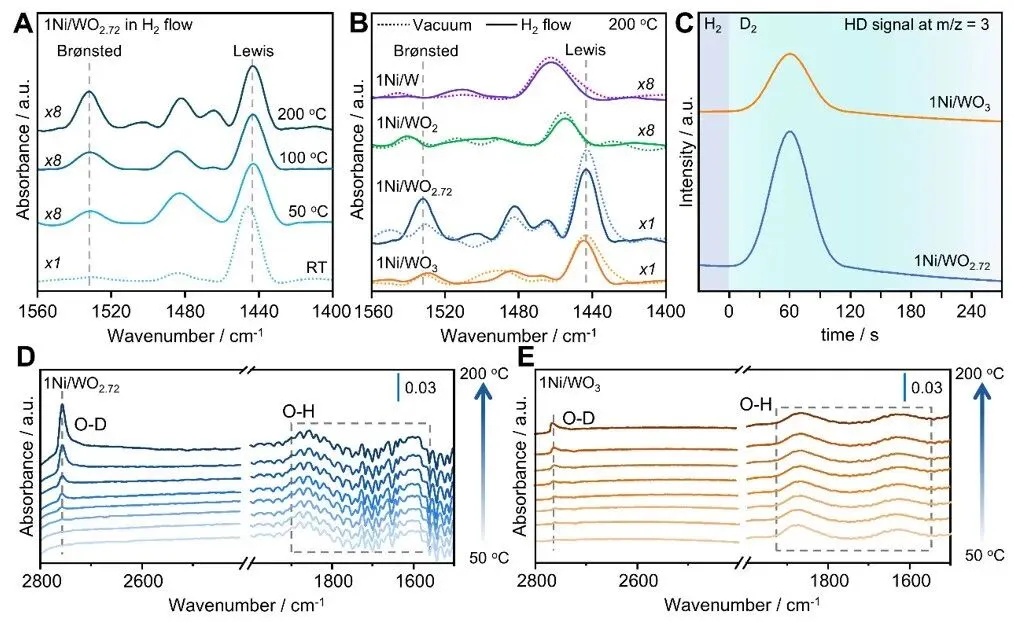
图3. 氢气活化与溢流。(A)不同温度下H₂气流中1Ni/WO₂.₇₂的原位吡啶红外光谱(Py-IR);(B)200℃下真空或H₂气流中1Ni/WOx样品的原位Py-IR光谱;(C)260℃ H-D交换实验中m/z = 3处的HD信号。(D)1Ni/WO₂.₇₂与(E)1Ni/WO₃在D₂气流中的原位傅里叶变换红外光谱(FT-IR)。
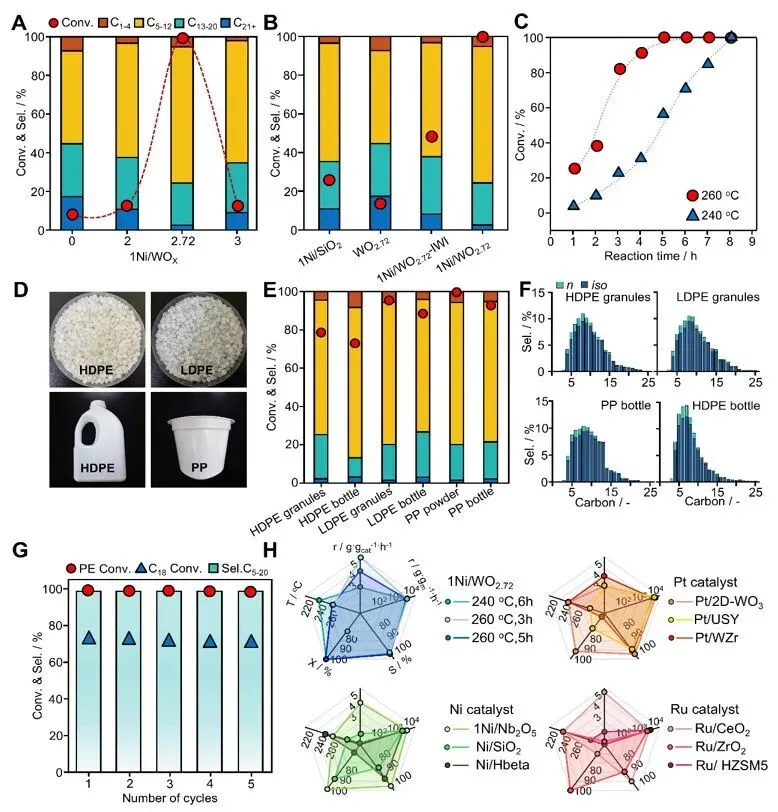
图4. 催化加氢裂化性能。(A)1Ni/WOx催化剂与(B)参照催化剂在PE加氢裂化中的催化性能;(C)1Ni/WO₂.₇₂在不同温度和反应时间下的催化性能;(D)真实及废弃塑料原料(包括颗粒和瓶体)实物图;(E)1Ni/WO₂.₇₂对不同塑料的转化率及产物分布;(F)不同塑料加氢裂化的详细产物分布;(G)1Ni/WO₂.₇₂在转化PE和C₁₈过程中的稳定性与可重复使用性;(H)1Ni/WO₂.₇₂与各类催化剂的液体燃料生成速率对比,详细数据汇总于表S10。反应条件:底物4.0 g;1Ni/WOx催化剂0.2 g;温度260℃;氢气压力3 MPa;反应时间5 h或按需注明。
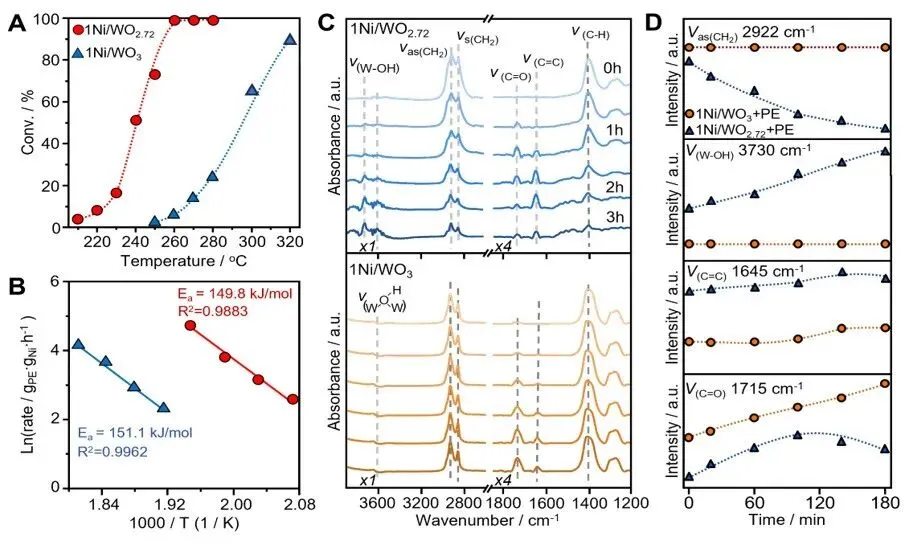
图5. 机理研究。(A)1Ni/WO₂.₇₂与1Ni/WO₃在不同温度下对聚乙烯(PE)的转化率;(B)1Ni/WO₂.₇₂与1Ni/WO₃催化PE加氢裂化的表观活化能(Ea);(C)260℃下1Ni/WO₂.₇₂与1Ni/WO₃催化PE加氢裂化的原位傅里叶变换红外光谱(FTIR);(D)表面物种的时间分辨演化:ν(CH₂)、ν(O–H)、ν(C═C)和ν(C═O)。
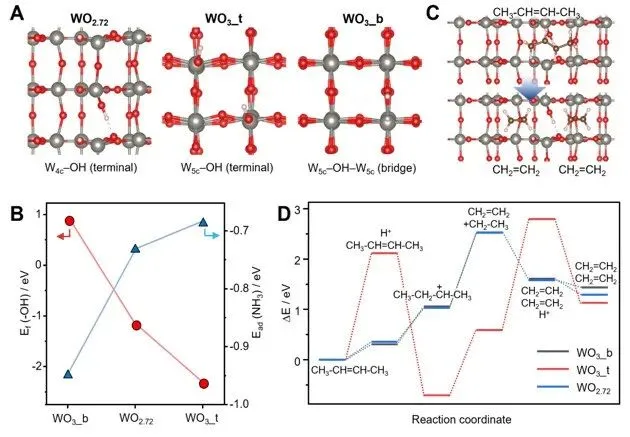
图6. 理论建模与反应路径计算。(A)WOx的布朗斯台德酸位点。从左至右依次为WO₂.₇₂、WO₃_t和WO₃_b表面的俯视图。(B)羟基的形成能及氨的解离吸附能。(C)WO₂.₇₂表面CH₃–CH═CH–CH₃裂解为CH₂═CH₂的瞬态图像。(D)丁烯在WOx表面加氢裂化的势能面起伏图。能量值以孤立丁烯与固体表面势能之和为参考基准。
通过将单原子镍(Ni)锚定在结构工程化的WO₂.₇₂纳米线上,我们实现了镍-氧-钨(Ni–O–W)位点上高效氢气(H₂)活化与溢流,进而促使氧空位处末端W═O基团转化为布朗斯台德酸性W–OH物种。所得1Ni/WO₂.₇₂催化剂在240–260℃下可高效、完全转化聚乙烯(PE),汽油-柴油燃料选择性达94.3%,且具有高生产率(5.0 g液体燃料/(g催化剂·h)),性能优于传统体系。原位光谱分析与密度泛函理论(DFT)计算表明,动态形成的末端W–OH基团(而非桥连W–OH-W)对激活涉及连续脱氢、质子化和C–C键断裂的加氢裂化路径至关重要。对比研究证实,C–C键断裂为速率决定步骤,且1Ni/WO₂.₇₂比1Ni/WO₃更易促进酸性位点形成。这些结果证明,催化位点的理性纳米结构设计对释放非贵金属的全部潜力,以及设计用于塑料升级转化及更广泛碳循环应用的高效、低成本催化剂至关重要。