10秒出结果!南京理工新模型TDFold让蛋白结构预测进入“平民算力”时代
- 2026-05-09 16:12:19
Two-dimensional geometric template diffusion for boosting single-sequence protein structure prediction
南京理工大学、北京师范大学、东南大学

近期推送
原文摘要
由于同源信息获取过程中计算成本高昂,基于单序列的蛋白质结构预测正受到越来越多的关注。本文提出一种名为TDFold的二维几何模板扩散方法,用于生成高质量的成对几何信息(包括成对距离与取向),并将其后续用于精准高效的三维蛋白质结构预测。针对给定的蛋白质序列,TDFold通过包含两个阶段的网络架构推断三维结构:二维几何模板生成阶段与序列-几何协同学习阶段。相较于现有蛋白质语言模型(如ESMFold、OmegaFold)及基于同源性的方法(如AlphaFold2、AlphaFold3与RoseTTAFold),TDFold具备三大核心优势:基于单序列的预测性能更优、资源消耗更低、推理效率更高。本研究在孤儿蛋白(Orphan)、Orphan25等同源信息匮乏数据集以及常用的CASP基准测试集上验证了模型有效性,为单序列蛋白质结构预测提供了一种新的解决方案,同时也加速了相关蛋白质研究进程,尤其适用于资源有限的高校与科研机构。
原文解读
研究背景
研究问题:
这篇文章旨在解决单序列蛋白质结构预测中的挑战,特别是当同源信息(如多个序列比对和三维结构模板)不足时,如何提高预测的准确性和效率。
研究难点:
同源信息依赖性:现有的深度模型高度依赖同源信息,导致在缺乏同源信息的蛋白质(如孤儿蛋白或快速进化的病毒蛋白)上预测准确性显著下降。
计算资源消耗:基于蛋白质语言模型的方法虽然不依赖同源信息,但通常采用大规模架构,计算资源消耗大,尤其是对于长序列蛋白质。
相关工作:
同源信息依赖方法:如AlphaFold2、AlphaFold3和RoseTTAFold,这些方法在CASP14数据集上表现出色,但依赖于同源信息,导致在孤儿蛋白等数据集上性能下降。
基于蛋白质语言模型的方法:如ESMFold、OmegaFold、RGN2和trRosettaX-single,这些方法不依赖同源信息,但采用大规模架构,计算资源消耗大。
研究方法
本文提出了一种名为TDFold的新框架,通过引入视觉生成模型来增强单序列蛋白质结构预测。具体方法如下:
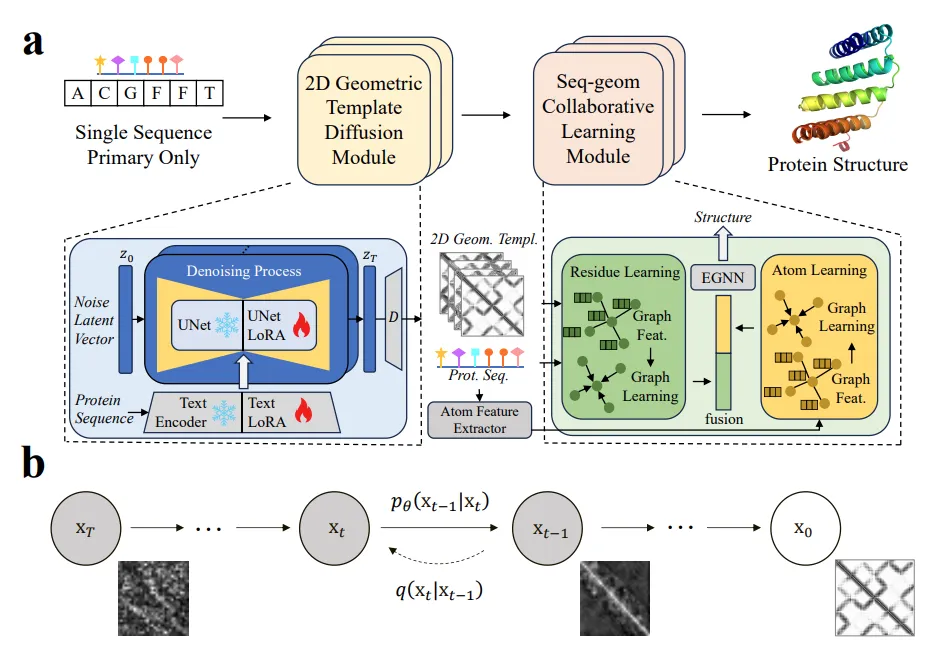
二维几何模板扩散模块:
扩散过程:通过概率扩散方法从蛋白质序列生成高质量的残基间几何信息(如距离和方向矩阵)。该过程包括一个前向过程(添加高斯噪声)和一个反向过程(逐步去噪以恢复几何图像)。
LoRA微调技术:对稳定扩散(SD)模型进行低秩适应(LoRA)微调,冻结原始训练参数,仅训练LoRA参数。通过文本LoRA和UNet LoRA将蛋白质序列和残基间几何图像特征映射到共享潜在空间,增强模型学习残基间几何信息的能力。
序列-几何协同学习网络(SCL):
残基级学习分支:通过图神经网络(GNN)学习残基间的序列和几何关系,构建残基图,传播信息以学习残基表示。
原子级学习分支:构建原子图,通过原子GNN处理原子和键的信息,学习包含侧链原子影响的表示。
特征融合与预测:通过变分学习框架融合残基级和原子级特征,注入侧链意识,最终通过SE(3)等变图神经网络(EGNN)预测全原子三维结构。
实验设计
数据集:
训练数据:来自PDB数据库,包含352,409个非冗余蛋白质域。
评估数据集:包括CASP14、CASP15、CASP16、Orphan和Orphan25数据集。Orphan和Orphan25数据集包含有限或无同源信息。
评估指标:
TM-score、GDT_TS和pLDDT用于评估蛋白质结构预测的性能。
实验设置:
二维几何模板扩散模块:将连续的残基间几何信息离散化为图像像素值范围(0-255),并通过LoRA微调SD模型生成高质量的残基间几何图像。
SCL网络:通过残基级和原子级学习分支提取特征,融合后预测三维结构。
结果与分析
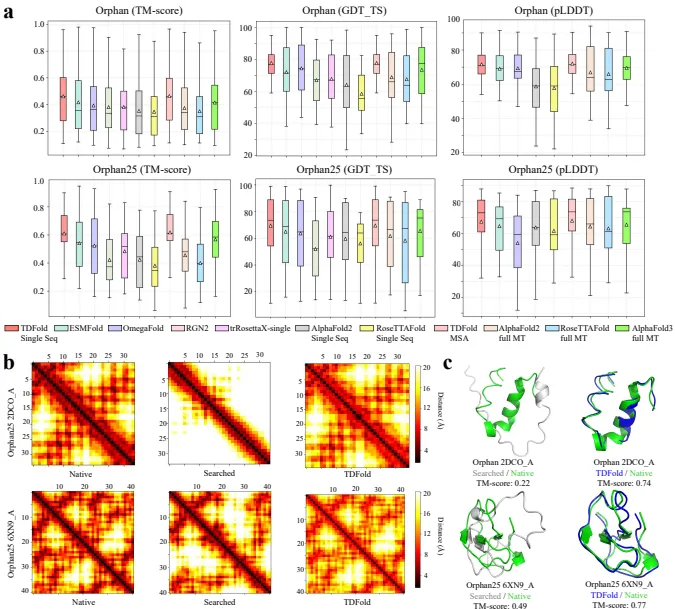
孤儿蛋白预测:
在Orphan和Orphan25数据集上,TDFold在TM-score、GDT_TS和pLDDT指标上均优于现有方法,如AlphaFold2、AlphaFold3、RoseTTAFold、ESMFold和OmegaFold。
CASP14、CASP15和CASP16数据集:
在CASP14、CASP15和CASP16数据集上,TDFold在单序列模式下表现出色,TM-score、GDT_TS和pLDDT指标优于ESMFold和OmegaFold,与AlphaFold2和RoseTTAFold相当。
计算效率:
TDFold在推理时间和GPU内存使用上显著优于现有方法。对于500个残基的蛋白质,TDFold的预测时间约为10秒,而AlphaFold2、AlphaFold3和RoseTTAFold分别需要约1000秒、240秒和100秒。GPU内存使用方面,TDFold约为7 GB,而AlphaFold2、RoseTTAFold和ESMFold分别为12 GB、16 GB和20 GB。
总体结论
本文提出的TDFold框架通过引入视觉生成模型,显著提高了单序列蛋白质结构预测的准确性和效率。TDFold在多个数据集上表现出色,特别是在缺乏同源信息的孤儿蛋白和快速进化的病毒蛋白上。此外,TDFold在计算资源消耗和推理时间上也具有显著优势,适用于大规模蛋白质结构预测任务。
特点评价
单序列预测性能优越:TDFold通过利用强大的文本到图像生成能力,使用真实的残基间几何信息作为监督信息,生成了高质量的残基间几何特征。这种方法消除了对同源数据的依赖,显著提高了单序列蛋白质结构预测的性能。
低资源消耗:通过采用轻量级的SCL架构,TDFold的GPU内存消耗通常低于流行的方法如AlphaFold2和ESMFold。
训练和推理效率更高:TDFold可以在一周内使用单个NVIDIA 4090 GPU完成训练,包括微调SD模型和从头开始训练SCL网络。此外,TDFold的推理时间显著短于现有方法(例如,对于长序列蛋白质,推理时间约为AlphaFold2和AlphaFold3的10倍到100倍)。
适用于资源有限的环境:TDFold可以在标准个人计算机上部署,使用单个NVIDIA 4090 GPU进行高效训练和推理,这对于资源有限的大学和学术机构特别有用。
大规模预测任务的适用性:TDFold在推理时间方面具有巨大优势,更适合需要高预测速度的大规模预测任务。

记录AI蛋白质设计在诺奖背后的人和事


